
Синдром тричера коллинза реферат
Обновлено: 19.05.2024
Диана — медсестра из Амурской области. На свой день рождения она родила сына Марка, ей исполнилось 28 лет. Беременность была тяжелой, поэтому решили делать плановое кесарево под общим наркозом.
Синдром Тричера Коллинза — аутосомно-доминантное заболевание, результат мутации в гене TCOF1. Характеризуется черепно-лицевой деформацией. По статистике, с ним рождается один ребенок из пятидесяти тысяч. Синдром описал английский офтальмолог Эдвард Тричер Коллинз в 1900 году.
У Дианы уже был четырехлетний сын Артем — абсолютно здоровый мальчик. Генетических заболеваний в семье нет. О том, что у второго сына возможен синдром, родители не подозревали. Потом врачи скажут Диане, что мутация случилась спонтанно.
Через четыре часа Диане разрешили встать и повели ее к Марку.
— Он лежал в кувезе: маленький, голый. Казалось, что там так холодно, а его даже не укрыли. Аппарат ИВЛ. Все это пикает… И внешне Марк был настолько для меня другим… не как обычные дети. Вернее, тело такое же, а на лицо — совсем другой.
При виде сына Диана потеряла сознание. Ее увели под руки, ребенка больше не показывали.
Марка забрали в областную больницу, а его мама осталась в отделении на тринадцать дней. В одиночестве ей стало страшно: казалось, что жизнь закончена.
— Я думала, что теперь засяду дома, никогда не выйду на работу и даже просто на улицу. В 28 лет можно было поставить огромный крест на карьере, хобби и любом развитии, потому что с ребенком надо будет находиться круглые сутки.
Из областной больницы Диане позвонила знакомая и рассказала, что мальчик жив: пока везли, он пытался дышать наперебой аппарату. Комплексное обследование показало, что внутренние органы не повреждены. Единственная проблема — Марк не мог дышать из-за дефекта лица.
На аппарате мальчик провел четыре дня, ему сделали первую операцию, чтобы открыть хоаны.
Ребенок самостоятельно дышал дня два, а после хрящи затянулись — и Марка снова положили на ИВЛ.
Когда Диану выписали, они с мужем смогли навещать сына почти каждый день. Их пускали в реанимацию, давали покормить мальчика через зонд, один раз даже разрешили подержать на руках.
За полтора месяца Марк привык к аппарату ИВЛ, но врач, который наблюдал ребенка с первого дня реанимации, сказал отключать и ставить трахеостому. Диана легла с сыном в отделение патологии, они провели там еще полтора месяца.
— Да, я видела людей с трахеостомой, я умею с ней работать по своей профессии. Но именно с ребенком это было страшно. Я не знала, как облегчить ему жизнь, какие операции мы можем сделать, чтобы вообще избавиться от этих трубок? Таня была первой, кто мне помог.
Они с Евгением уставали, у кроватки приходилось сидеть круглые сутки — менялись по часам, чтобы сын не оставался без присмотра даже ночью.
— А меня пугала сама мысль, что мой ребенок может остаться один, в изоляторе… В доме малютки никто бы за ним так не смотрел, и Марк бы просто задохнулся. Мы с мужем обсуждали, как бы мы жили без него, и поняли — не сможем. Как жить спокойно, сидя на диване и попивая чаек, когда где-то мучается твой никому не нужный ребенок?
Все знакомые семьи говорят, что Марк, несмотря на синдром, очень похож на папу, и Евгений этим очень гордится.
Через три месяца после рождения Марка наконец выписали домой. Из-за гипоксии он остался лежачим. Особенно тяжело родителям было по ночам: приходилось смотреть за сыном, и они спали по два–три часа.
— Я боялась, что не буду его слышать, допустим, ночью, если он заплачет. Как говорят, дети с трахеостомой — шипящие дети. У них плач — шипение, смех — шипение. Но я до сих пор слышу все, как бы ни уставала. Даже если он на другом конце комнаты: когда просыпается, начинает кашлять, переворачивается.
Диана по-прежнему не могла смириться с тем, что ее жизнь навсегда изменилась. Когда было совсем трудно, Евгений заменял сыну и маму, и папу.
— Первые полгода я плакала и жалела себя. Было полное отрешение от мира и даже от собственной семьи. Жить не хотелось точно. Но я понимала, что нужно — и ради Марка, и ради старшего сына.
Диане нужно было рано вставать, кормить Артема, отводить его в садик, стирать, убирать. Она старалась, чтобы сын не чувствовал ее горя и так же, как раньше, ходил на кружки и секции, так же вкусно ел и играл с другими детьми. Теперь она понимает, что именно Артем помог ей выбраться из депрессии: ради него вставала, шла и делала.
Следующая операция была в Москве, после нее мальчик долго не поднимал руку, брал игрушку и из-за тремора не мог ее удержать.
— Для меня это было очень дико.
Мы первое время сравнивали наш менталитет и менталитет людей за границей. Там в моего ребенка ни разу не тыкнули пальцем.
Наоборот, подходили, теребили за щечки и были готовы расцеловать. В магазине нам совали фрукты, еще что-то, двери открывали, просто улыбались.
Когда прилетели в Москву, Диана боялась показывать Марка и закрывала его лицо — казалось, что люди смотрят брезгливо. Девять часов она просидела в комнате матери и ребенка, чтобы их просто никто не видел.
Со временем агрессии стало меньше. Сегодня в области Диану хорошо знают. Когда, например, звонит в минздрав или в хирургическое отделение, достаточно сказать, что она мама ребенка с синдромом Тричера Коллинза.
Ползает — но на спине, как лягушка
Сейчас Марку два с половиной года. Родители возят его на реабилитации, занимаются с ним дома. Деньгами на ходунки и другие приспособления помогают люди.
— Так вот и обходимся, — говорит Диана. — То ли живем, то ли существуем, пытаемся поставить Марка на ноги.
И хотя врачи говорили, что мальчик не встанет, полгода назад он научился присаживаться и вставать на коленки. Ползает — но на спине, как лягушка. Очень активный, достает из ящиков все, что можно и нельзя.
У Марка два слуховых аппарата костной проводимости, он их носит на резинке, иногда пытается снять. Маме кажется, что он слышит громкие звуки и без аппаратов: когда старший сын бегает, прыгает и пищит, Марк смеется и смотрит в ту сторону. Но когда родители стоят и что-то говорят за спиной, он не обращает внимания: то ли ему неинтересно, то ли просто не слышит.
Игрушки Марк любит музыкальные, каждую прикладывает к уху и проверяет, играет она или нет. Мультики смотрит в аппаратах. Если не нравится — закрывает руками глаза.
Как только Марк начнет самостоятельно дышать, ему уберут трахеостому. После этого он будет учиться есть. У него уже появился сосательно-глотательный рефлекс.
— Кроме этого, как нам объяснил невролог, есть возможность, что функцию погибшего участка мозга возьмут здоровые клетки, и все восстановится. Конечно, Марк не сможет активно бегать и прыгать, но обслуживать себя должен научиться. Пока что по сравнению со всеми Тричерами, которых я встречала, нам досталось немало: к трудностям синдрома добавилась еще и неврология.
— Это было настолько обидно! Я не сделала никому плохого. Мне было 28 лет, я работала в медицине, спасала людей. Что я могла такого совершить? Но в этом состоянии начинаешь копаться в себе и правда искать, где же ты что-то натворила.
— И вы знаете, после этих слов я начала жить. Значит, все, что мне дано, мне по силам. И эти мысли, что ребенок со мной действительно ни в чем не будет нуждаться, меня успокаивают.
— На работе голова отдыхает. Я понимаю, что есть не только мои проблемы с сыном, отвлекаюсь, и это мне помогает не углубиться в уныние дальше.
— Если мне куда-то нужно, я и накрашусь, и волосы выпрямлю, и наряжусь. Но по будням до такой степени устаю физически, что на эти вещи нет сил. К тому же это тяжело морально. Проблемы все равно не отпускают. Я учусь с ними жить, в то же время мне очень трудно совмещать и работу, и семейные дела. Сейчас мечта — просто выспаться.
Лидер продаж в высоком классе. Монитор 21,5" высокой четкости, расширенный кардио пакет (Strain+, Stress Echo), экспертные возможности для 3D УЗИ в акушерско-гинекологической практике (STIC, Crystal Vue, 5D Follicle), датчики высокой плотности.
Синдром Тричера Коллинза (СТК, Treacher Collins syndrome) – это врожденное, наследственно обусловленное нарушение развития производных первой глоточной дуги, которое характеризуется специфическими черепно-лицевыми проявлениями: двусторонней симметричной отонижнечелюстной дисплазией с гипоплазией скуловых костей.
Синонимы: синдром Франческетти, синдром Тричера Коллинза–Франческетти, синдром Франческетти–Цвалена–Клейна, челюстно-лицевой дизостоз.
Отличительные признаки СТК: гипоплазия скуловых костей, антимонголоидный разрез глаз, опущенные уголки глаз, колобома нижнего века, пороки развития наружного уха [1, 2].
Хотя первым болезнь описал Аллен Томсон еще в 1846 г., синдром обычно называют именем врача Тричера Коллинза, который в 1900 г. описал двух больных с похожими симптомами. Не верно писать этот синдром через дефис, так как Тричер – имя доктора Коллинза. Уже в 40-х годах прошлого века Адольф Франческетти и Давид Клейн дали подробную характеристику болезни и назвали ее челюстно-лицевым дизостозом [3]. В некоторых странах Европы этот синдром называют синдромом Франческетти или синдромом Тричера Коллинза–Франческетти [4, 5].
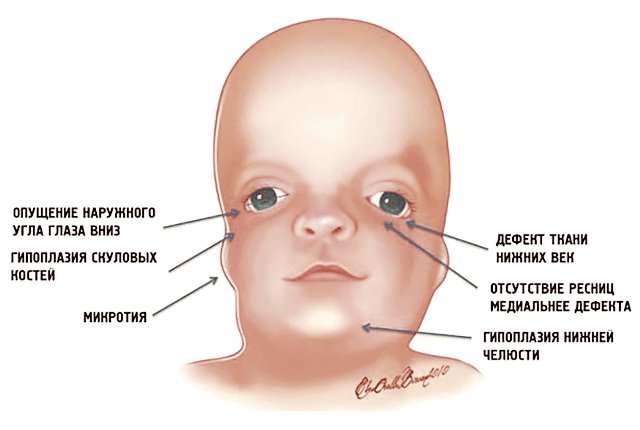
Рис. 1. Схема специфических признаков лицевых дизморфий при синдроме Тричера Коллинза.
Наиболее частые симптомы и фенотипические проявления СТК
У людей с СТК отмечается характерный лицевой дизморфизм (рис. 1) с двусторонней симметричной гипоплазией скуловых костей (95%), характерна гипоплазия инфраорбитального края глазницы (80%) с формированием антимонголоидного разреза глаз (89%) и гипоплазией нижней челюсти (78%), что приводит к аномалии прикуса [1–6], также наблюдается апертогнатия (так называемый открытый прикус). Описана атрезия хоан [7], колобома (расщелина) нижних век между внешней и средней третью (69%), сопровождающаяся отсутствием ресниц. Гипоплазия мягких тканей преимущественно отмечается в скуловой области, нижнем орбитальном крае и щеках. К особенностям относятся сложные нарушения в строении височно-нижнечелюстного сустава, что приводит к ограниченной воз можности открытия рта различной степени тяжести [1].
Часто отмечается аномалия наружного уха, например микротия или анотия (77%), атрезия наружного слухового прохода и аномалии развития слуховых костей (60%), что приводит к кондуктивной тугоухости [1–8]. Снижение зрения, вплоть до полной его потери, встречается в 37% случаев. Нёбо высокое, имеет готическую форму и иногда наблюдается его расщелина (28%).
Умственные способности, как правило, нормальные. Умственная отсталость встречается лишь у 5% людей с СТК [1, 2]. Из-за узких верхних дыхательных путей и ограниченного открывания рта в раннем возрасте могут возникать трудности с дыханием и питанием [8]. Из частых признаков описан чрезмерный рост волос на щеках [2, 8, 9].
Этиология синдрома Тричера Коллинза
На сегодняшний день описано три типа СТК. До 93% всех случаев – это синдром 1-го типа [10]. СТК 1-го типа связан с мутациями гена TCOF1, который расположен в сегменте 5q32 – q33. Тип наследования аутосомно-доминантный [2] с 90% пенетрантностью и переменной экспрессивностью (проявляемостью), даже у пациентов в пределах одной семьи. Известны наблюдения детей с выраженными клиническими проявлениями синдрома в одной семье, тогда как у одного из их родителей была обнаружена та же мутация без выраженных клинических проявлений болезни [2, 4–6]. Около 60% случаев СТК не наследуются от больных родителей, а являются новыми мутациями (de novo).
Также описаны 2-й и 3-й типы СТК. Второй тип вызван мутацией гена POLR1D на хромосоме 13q12, 3-й тип – мутацией гена POLR1C на хромосоме 6p21. Нужно отметить, что клинически все три типа не отличаются друг от друга, несмотря на то что мутации затрагивают разные гены, на разных хромосомах [2] и тип наследования может быть и аутосомно-рецессивным [11].
Пренатальная диагностика СТК
Дифференциальная диагностика СТК должна включать некоторые генетические синдромы с преимущественным поражением лицевых структур [17]:
- Синдром Гольденхара. Изменения лица при синдроме Гольденхара почти всегда односторонние, асимметричные, включают в себя колобому верхнего, а не нижнего века, а также эпибульбарные дермоиды, преаурикулярные привески. При синдроме Гольденхара могут встречаться аномалии позвоночника и пороки сердца.
- Синдром Нагера. Фенотипически похож на СТК, однако для него характерны преаксиальные (со стороны большого пальца кисти) дефекты верхней конечности – редукционные пороки верхних конечностей (в диапазоне от гипоплазии до аплазии большого пальца с или без вовлечения лучевой кости).
- Синдром Миллера, известный как постаксиальный акрофациальный дизостоз. Характеризуется микрогнатией, расщелиной губы, различными аномалиями позвонков и сколиозом. Типичными признаками являются постаксиальные (со стороны мизинца кисти) пороки верхней конечности либо только мизинца.
- Синдром Пьера Робена характеризуется изолированной гипоплазией нижней челюсти, глоссоптозом, расщелиной нёба.
Следует подчеркнуть, что аномалии конечностей не свойственны для СТК и для синдрома Пьера Робена, и, если они присутствуют, следует больше думать о синдромах Миллера или Нагера.
Профилактика и лечение СТК
Генетическое консультирование семей с больным ребенком/плодом осложняется вариабельной проявляемостью заболевания и должно осуществляться мультидисциплинарной группой специалистов по пренатальной диагностике с обязательным выяснением этиологии возникновения заболевания в конкретной ситуации (семейная форма либо мутация de novo). При наличии у родителя признаков СКР единственным эффективным методом профилактики заболевания следует назвать применение методик экстракорпорального оплодотворения с предимплантационной диагностикой с целью переноса здоровых эмбрионов, либо применение донорских ооцитов или сперматозоидов.
При продолжающейся беременности послеродовое ведение требует междисциплинарного подхода (акушер, неонатолог, хирург, анестезиолог и генетик); и из-за возможных острых проблем с дыханием роды должны планироваться в специализированных перинатальных центрах. Лечение больных с СТК многопрофильное. В случае возникновения постнатального респираторного дистресс-синдрома необходимо применение трахеостомии, неинвазивной вентиляции и дистракции нижней челюсти. Челюстно-лицевая и пластическая хирургия позволяет устранить гипоплазию мягких тканей (коррекция овала лица с помощью липоскульптуры), гипоплазию костной ткани (хирургическая дистракция кости, костные трансплантаты), колобому век и расщелину нёба (хирургическое восстановление). Для устранения аномалий среднего уха (функциональная хирургия) и наружного уха (реконструкция ушных раковин) требуется участие специалиста в области ЛОР-хирургии. Коррекция нарушения слуха должна осуществляться на ранней стадии (слуховые аппараты и функциональная хирургия), что способствует нормальному развитию ребенка.
При надлежащем лечении прогноз для легких форм заболевания является благоприятным. Для тяжелых форм заболевания с выраженными клиническими проявлениями прогноз неблагоприятный не только для здоровья, но и для жизни.
Описание случая синдрома Тричера Коллинза
В медико-генетическом отделении (МГО) Московского областного НИИ акушерства и гинекологии для консультации по прогнозу потомства и возможностях обследования обратилась пациентка 25 лет со сроком беременности 8 нед. Данная беременность вторая. Брак не родственный. Муж здоров, производственных вредностей супруги не имеют. Первая беременность закончилась преждевременными родами в сроке 36 нед. Родилась девочка с массой тела 1990 г, ростом 51 см, с оценкой по шкале Апгар 7/7 баллов. При осмотре ребенка генетиком выявлены особенности фенотипа, характерные для СТК: гипоплазия скуловых костей, антимонголоидный разрез глаз, гипоплазия нижней челюсти, двусторонняя микротия с атрезией слуховых проходов. Методом автоматического прямого секвенирования был проведен поиск мутаций в гене TCOF1. Выявлен патогенный вариант c.3946_3947 delGA в гетерозиготном состоянии. Ребенку выставлен клинический диагноз: синдром Тричера Коллинза. Тяжесть состояния ребенка усугубилась врожденной пневмонией, церебральной ишемией II степени, недоношенностью, анемией тяжелой степени. Ребенок был переведен в отделение реанимации, умер в 1,5 мес. При консультировании ребенка генетиком риск повторного рождения больного ребенка в семье расценен как низкий, так как данная мутация расценена генетиком как мутация de novo. Дана рекомендация о пренатальной диагностике и кариотипировании плода при следующей беременности без указания на необходимость специфической диагностики СТК. Пациентка самостоятельно обратилась для обследования в медико-генетический научный центр (МГНЦ). В образце ее ДНК методом прямого автоматического секвенирования была найдена патогенная мутация в гене TCOF1 в гетерозиготном состоянии. Таким образом, у пациентки тоже имеется СТК и риск рождения у нее больных детей будет высоким – 50%. При предыдущем осмотре генетиком ее фенотип был не изучен и не оценен в полном объеме. При внимательном осмотре пациентки найдены мягкие, но классические признаки СТК: опущенные уголки глаз, колобомы нижнего века, рост волос на лице, гипоплазия мягких тканей в области скуловых дуг. При сборе анамнеза выяснено, что пациентка страдает двусторонней тугоухостью. С учетом аутосомно-доминантного типа наследования СТК, известного картированного патологического гена было рекомендовано проведение инвазивной пренатальной диагностики с прицельным поиском известной мутации и экспертное ультразвуковое исследование в 12–13 нед беременности.
При ультразвуковом исследовании выявлены множественные особенности лицевого фенотипа у плода: микрогнатия (рис. 2–4), треугольная форма лица (рис. 5), опущенные книзу глазницы и гипоплазия скуловых дуг (рис. 6, 7), аномальная форма и положение ушей (рис. 5, 7).
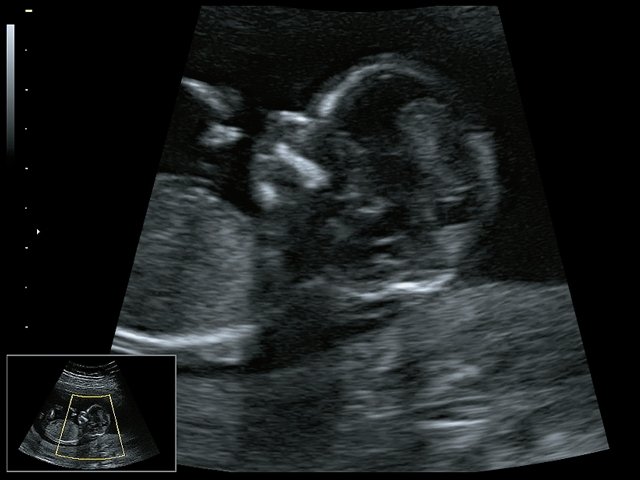
Рис. 2. Микрогнатия - сагиттальный скан в 2D, беременность 13 нед.
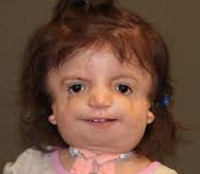
Синдром Тричера Коллинза – это генетическая (иногда наследственная) болезнь, сопровождающаяся деформациями костей и мягких тканей лица. К симптомам относятся грубые дефекты строения лица: антимонголоидный разрез глаз, вырезки ткани век (колобомы), уменьшенные размеры челюсти и скул, гипоплазия и аномалии структур уха, расщелина или арковидная форма неба, увеличенные размеры ротовой щели и языка, слаборазвитые кости лица. Диагноз устанавливается по данным клинического осмотра, биогенетического теста и семейного анамнеза. Лечение симптоматическое, направлено на улучшение слуха, устранение жизнеугрожающих деформаций и косметических дефектов хирургическим способом.
МКБ-10
Общие сведения
Причины
При мутациях в гене TCOF1 тип наследования синдрома аутосомно-доминантный с показателем пенетрантности 90%. Это означает, что при мутации в одной хромосоме из пары вероятность проявления болезни очень высока. У больного родителя риск рождения ребенка с синдромом Тричера Коллинза составляет 50%. Возможна наследственная передача дефекта и спорадические генетические изменения (новые мутации). Экспрессивность мутации переменная – в пределах одной семьи вероятно как ослабление, так и усиление симптомов заболевания у последующих поколений. При дефектах генов POLR1C и POLR1D наследование происходит по аутосомно-рецессивному типу. В парах, где родитель имеет синдром, вероятность рождения больного малыша составляет 25%.
Патогенез
Пятая хромосома ответственна за правильное формирование скелета в период внутриутробного развития. Локализованный в ней ген TCOF1 кодирует структуру и синтез ядерного транспортного белка Treacle. Данный протеин экспрессируется в большинстве тканей организма в эмбриональном и постэмбриональном периоде, участвует в переносе генетической информации с ДНК на РНК.
В основе синдрома чаще всего лежит нонсенс-мутация, приводящая к образованию преждевременного кодона терминации и развитию гаплонедостаточности – дефицита белка, необходимого для нормального формирования лицевой части черепа. Здоровый ген обеспечивает организм белком Treacle наполовину, но такого количества недостаточно для правильного развития лицевых структур. При изменениях в генах POLR1D и POLR1C процесс транскрипции ДНК нарушается из-за недостаточности фермента-катализатора ДНК-зависимой РНК-полимеразы. Клинические проявления синдрома такие же, как и при первичной недостаточности Treacle-протеина.
Симптомы
У 69% пациентов определяется колобома радужки и нижних век в промежутке между средней и внешней третью, чаще она имеет треугольную форму. Ресницы на внешнем крае нижнего века отсутствуют. Небо арковидной формы, иногда сформирована расщелина (у 28% больных). Аномалии наружного уха представлены недоразвитием или полным отсутствием ушной раковины (микротией, анотией), атрезией наружного слухового прохода и деформацией слуховых косточек. Зачастую пациенты имеют кондуктивную тугоухость. В редких случаях диагностируется энхондрома, предкозелковые фистулы, аномальное строение сердца и позвоночника.
Осложнения
Микрогнатия и стеноз верхних дыхательных путей уже в первые годы жизни могут спровоцировать проблемы при приеме пищи и трудности дыхания вплоть до удушья. Своевременная диагностика заболевания позволяет спрогнозировать эти осложнения и предпринять меры по их предупреждению. Как правило, пациенты не имеют врожденных интеллектуальных расстройств, но при отсутствии коррекции нарушений слуха становится невозможным правильное формирование речи и обучение в обычных условиях. Дети начинают отставать в умственном развитии от сверстников, имеют задержку психического развития различной степени выраженности. В связи с наличием дефектов внешности и негативным отношением окружающих больные всех возрастов относятся к группе риска по возникновению депрессии, ипохондрии, тревожности и иных невротических расстройств.
Диагностика
Диагноз может быть установлен во время беременности или сразу после рождения. Обследование показано женщинам из группы риска и детям с врожденными лицевыми деформациями. В процессе диагностики принимают участие врачи-генетики и педиатры. Синдром Тричера-Коллинза необходимо дифференцировать с другими генетическими заболеваниями, при которых существует деформация лицевой части черепа, например, с синдромом Нагера и синдромом Гольденхара. Используются следующие методы:
Дополнительно назначаются обследования, позволяющие своевременно обнаружить жизнеугрожающие состояния, оценить степень деформации костей черепа. Определяется эффективность кормления ребенка, уровень насыщения гемоглобина кислородом, ритмичность и глубина дыхания. Для диагностики сохранности слуха на 5-6 день жизни проводится аудиологическое тестирование. Инструментальная диагностика включает рентгенографию черепа, КТ и МРТ головного мозга.
Лечение синдрома
Специфической терапии не существует. Лечение нацелено на устранение симптомов и последствий заболевания, предполагает проведение хирургических операций и реабилитационных мероприятий. Объем процедур и сроки их выполнения устанавливаются индивидуально с учетом наличия угрозы для жизни больного, противопоказаний и рисков, связанных с оперативным вмешательством. Общая схема лечения включает:
- Восстановление глотания и дыхания. При развитии респираторного дистресс-синдрома осуществляется трахеостомия, дистракция подвижной челюсти, неинвазивная вентиляция легких. При невозможности потребления пищи устанавливается гастростома.
- Восстановление слуха. Деформация наружного и среднего уха устраняется хирургическим путем, но потеря слуха чаще обусловлена повреждением слуховых мелких косточек, поэтому оперативные вмешательства с целью устранения тугоухости неэффективны. Предпочтительна реабилитация слуховыми аппаратами.
- Устранение внешних дефектов. Деформации корректируются методами пластической и нижнечелюстно-лицевой хирургии. Применяется липоскульптурирование, хирургическая дистракция костей, установка трансплантатов и хирургическое восстановление неба.
Прогноз и профилактика
Комплексное лечение и реабилитация значительно улучшают качество жизни больных. При легкой и умеренной выраженности синдрома прогноз благоприятный. Профилактика затруднена, поскольку заболевание является генетическим, а мутации способны возникать спонтанно. Супружеским парам, в которых один родитель болен, необходимо медико-генетическое консультирование и перинатальная диагностика синдрома на ранних сроках беременности. Для снижения риска вынашивания больного ребенка рекомендуется процедура экстракорпорального оплодотворения с предварительным отбором генетически здоровых эмбрионов.
1. Медицинская и клиническая генетика для стоматологов: учебник для вузов. Под ред. О. О. Янушевича – 2009.
2. Особенности стоматологической патологии при некоторых наследственных заболеваниях / Шишкова О.В., Максимова Ю.В. // Медицина и образование в Сибири – 2007 - №3.
Для заболевания характерна высокая пенетрантность (т.е. высокая вероятность проявления признаков болезни у людей с мутацией) и различная экспрессивность (т.е. различный характер и тяжесть проявления болезни). Молекулярно-генетической причиной заболевания являются, как правило, нонсенс мутации в генеTCOF1, приводящие к возникновению преждевременного стоп кодона и, как следствие, к гаплонедостаточности (состояние, при котором половинного количества генного продукта недостаточно для нормального функционирования организма). Продукт гена - ядерный транспортный белок, который экспрессируется во многих тканях во время эмбрионального и постэмбрионального развития и принимает участие в транскрипции ДНК. Возможна прямая молекулярно-генетическая диагностика этого синдрома, которая заключается в выявлении изменений нуклеотидной последовательности в гене TCOF1 методом прямого автоматического секвенирования.
Аутосомно-доминантный. Для заболевания характерна высокая пенетрантность (т.е. высокая вероятность проявления признаков болезни у людей с мутацией) и различная экспрессивность (т.е. различный характер и тяжесть проявления болезни).
Мутацию в гене TCOF1 можно выявить в 78%-93% случаев, в 8% выявляют мутации в генах POLR1C или POLR1D.
Продукт гена - ядерный транспортный белок, который экспрессируется во многих тканях во время эмбрионального и постэмбрионального развития и принимает участие в транскрипции ДНК. У больных весьма характерное лицо. Клинические признаки: антимонголоидный разрез глаз, колобома (дефект) нижних век, микрогнатия (гипоплазия нижней челюсти), двусторонняя гипоплазия скуловых костей и орбит, аномалии ушных раковин, дефект слухового прохода, приводящий к кондуктивной тугоухости. Часто встречается высокое арковидное небо или расщелина неба, макростомия (большая ротовая щель), отсутствие ресниц на нижнем веке. В некоторых случаях отмечаются рост волос на щеках, колобомы верхнего века и радужки. Порок развития уха заключается в деформации ушной раковины, отсутствии костного отдела наружного слухового прохода, недоразвитии барабанной полости и слуховых косточек. Отмечаются также гипоплазия больших пальцев лучевой и локтевой костей, расщелины неба. Тугоухость смешанного характера с одновременным поражением звукопроведения и звуковосприятия.
Интеллект, как правило, не страдает. Трудности с дыханием и питанием могут возникнуть в первые годы из-за стеноза верхних дыхательных путей и ограниченного открытия рта.
Литература
Специальной подготовки к исследованию не требуется.
Обязательны к заполнению:
ИНВИТРО гарантирует конфиденциальность и неразглашение предоставляемой пациентом информации в соответствии с законодательством Российской Федерации.
Литература
Типичная клиническая картина.
Литература
Интерпретация результатов исследования содержит информацию для лечащего врача и не является диагнозом. Информацию из этого раздела нельзя использовать для самодиагностики и самолечения. Точный диагноз ставит врач, используя как результаты данного обследования, так и нужную информацию из других источников: анамнеза, результатов других обследований и т.д.
синдром I-II жаберных дуг, гемифациальная микросомия, синдром Гольденхара, синдром Пьера Робена, синдромы Нагера.
Читайте также: