
Синдром коффина лоури реферат
Обновлено: 02.07.2024
Нарколепсия — расстройство сна с эпизодами непреодолимой дневной сонливости. Нарколепсии часто сопутствуют: нарушение сна ночью, катаплексия, сонный паралич, гипнагогические и гипнопомпические галлюцинации.
Катаплексия — вызванное сильными эмоциями внезапное снижение или потеря мышечного тонуса при сохранении сознания.
Сонный паралич — временная генерализованная неспособность двигаться или говорить при засыпании или пробуждении.
Гипнагогические и гипнопомпические галлюцинации — яркие галлюцинации соответственно в начале сна и при пробуждении.
Существует две формы нарколепсии:
- нарколепсия типа 1 (NT1), ранее называвшаяся нарколепсией с катаплексией, характеризуется наличием катаплексии, очень низким уровнем (≤ 110 пг/мл) гипокретина 1 (орексин A) в спинномозговой жидкости, тяжелой и высокоизбирательной потерей гипокретиновых нейронов в гипоталамусе;
- нарколепсия типа 2 (NT2), прежде называвшаяся нарколепсией без катаплексии, характеризуется отсутствием катаплексии, нормальным уровнем гипокретина 1 в спинномозговой жидкости, менее выраженной потерей гипокретиновых нейронов в гипоталамусе (у 10–30% пациентов).
Развитие нарколепсии типа 1 связано, как предполагается, с генетическими и внешними факторами, определяющими аутоиммунную атаку на гипокретиновые нейроны у генетически предрасположенных людей.
Патогенез нарколепсии типа 2 неизвестен.
Распространенность нарколепсии в мире укладывается в диапазон 0,025–0,05%. Нарколепсией обычно страдают дети, подростки и молодые люди. Пик заболевания приходится на второе десятилетие жизни. С момента появления симптомов нарколепсии до постановки ее диагноза обычно проходит 5–15 лет.
Пациенты с нарколепсией испытывают недостаток бдительности и устойчивого внимания, замедленное время реакции, повышенную частоту совершения ошибок, трудности с выполнением комплексных, монотонных и продолжительных задач.
Среди потенциальных осложнений нарколепсии: повышенные риски дорожно-транспортных происшествий (если оставлять ее без какого-либо лечения), травмы, переломов костей, падения (по причине катаплексии и/или эпизодов дневной сонливости). Нарколепсия также ассоциирована с повышенной смертностью.
Демография нарколепсии
Нарколепсии чаще всего подвержены дети, подростки и молодые люди: заболевание обычно проявляется в возрасте 10–20 лет, хотя манифестация болезни может произойти в любом возрасте.
С момента манифестации симптомов нарколепсии до постановки ее диагноза проходит 5–15 лет.
Второй пик случаев нарколепсии приходится на возраст 30–39 лет.
Заболеваемость и распространенность нарколепсии
Мировая распространенность нарколепсии укладывается в пределы 0,025–0,05%, хотя некоторые оценки базируются на опросниках пациентов, а не медицинских диагнозах.
Заболеваемость нарколепсией варьирует в зависимости от возрастной группы и географического региона в дополнение ко времени возникновения пандемии вируса гриппа A серотипа H1N1 — самого распространенного типа гриппа. Возможные объяснения включают различия в факторах генетического риска и воздействие факторов окружающей среды:
Факторы риска нарколепсии
Среди вероятных факторов риска нарколепсии: сочетание генетических факторов, возраста и определенных иммунных стимулов:
Генетика
- у родственников первой линии родства риск развития нарколепсии в течении жизни составляет 1–2%;
- коэффициент конкордантности у монозиготных близнецов равен 25–31%;
- аллель DQB1*0602 лейкоцитарного антигена человека (HLA) ассоциирован с нарколепсией и встречается у 88–98% пациентов с нарколепсией типа 1, у 40–60% страдающих нарколепсией типа 2, у 12–34% человек из общей популяции;
- нарколепсия связана с рисковыми аллелями дополнительных генов, участвующих в функционировании иммунной системы, как то:
- альфа-локус T-клеточного рецептора: отвечает за распознавание антигенов;
- P2RY11: рецептор, экспрессирующий на T-клетках CD8+;
- катепсин H: вовлечен в процессинг и презентацию антигенов на молекулах главного комплекса гистосовместимости;
- TNFSF4/OX40L: костимулирующий фактор для T-клеточной активации.
Возраст
Нарколепсия чаще всего поражает детей, подростков и молодых взрослых (10–20 лет), притом что в среднем проходит 5–15 лет между манифестацией симптомов заболевания и постановкой его диагноза.
Возраст манифестации нарколепсии характеризуются бимодальностью: первый пик болезни приходится на 15-летний возраст, второй — на 35-летний.
Одно крупное популяционное исследование указало на единственный пик нарколепсии между 10 и 19 годами — с последующим постепенным снижением вероятности.
Иммунные триггеры
Стрептококковые инфекции, возможно, ассоциированы с повышенным риском развития нарколепсии.
Этиология и патогенез нарколепсии
Этиология нарколепсии
Нарколепсия типа 1 вызывается тяжелой, но высокоизбирательной потерей продуцирующих гипокретин (орексин) нейронов в гипоталамусе.
Нарколепсия типа 2 может быть вызвана менее выраженным повреждением продуцирующих гипокретин нейронов в гипоталамусе, но свидетельствующие об этом данные ограничены.
Среди второстепенных причин нарколепсии:
- саркоидоз;
- рассеянный склероз и другие демиелинизирующие заболевания;
- генетические нейродегенеративные заболевания;
- инсульт;
- опухоли;
- агенез мозолистого тела;
- нейроцистицеркоз;
- травматическое повреждение головного мозга;
- лимбический энцефалит;
- паранеопластический синдром;
- синдром Гийена — Барре;
- врожденный порок развития головного мозга;
- болезнь Ниманна — Пика типа C;
- болезнь Норри;
- синдром Коффина — Лоури;
- синдром Прадера — Вилли;
- аутосомно-доминантная мозжечковая атаксия.
Патогенез нарколепсии
Точные патогенетические механизмы нарколепсии, особенно типа 2, неизвестны.
Развитие нарколепсии типа 1 связано, есть мнение, с генетическими и внешними факторами, которые, возможно, результируют аутоиммунными атаками на гипокретиновые нейроны у генетически предрасположенных людей.
Гипотеза отталкивается от сильной связи между нарколепсией типа 1 с аллелью DQB1*0602 лейкоцитарного антигена человека (HLA) и полиморфизмом других иммуносвязанных генов.
Гипотеза также опирается на определенную сезонность манифестации нарколепсии типа 1, что указывает на инфекционный триггер заболевания.
Среди других фактов в пользу аутоиммунной природы нарколепсии типа 1:
- отчеты о спорадической совместной встречаемости нарколепсии и рассеянного склероза — аутоиммунного заболевания, связанного с DQB1*0602;
- отчеты о связи между нарколепсией и аутоиммунными паранеопластическими синдромами, такими как лимбический энцефалит с антителами против Ma2;
- отчеты о частичном клиническом ответе пациентов с нарколепсией после назначения иммуноглобулина G;
- наличие аутореактивных антител и T-клеток в образцах крови пациентов с нарколепсией.
Гипокретиновая сигнализация усиливает активность в областях головного мозга, подавляющих фазу быстрого сна. Потеря гипокретиновой сигнализации отражается следующими проявлениями:
- противоречивой активностью в областях головного мозга, способствующих бодрствованию, что выражается частыми нарушениями сна;
- наличием во время бодрствования элементов фазы быстрого сна, таких как паралич или сонные галлюцинации.
Катаплексия, вызванная сильными эмоциями, передающимися через медиальную префронтальную кору и миндалевидное тело и активирующими цепи в мосте, приводит к мышечному параличу.
У почти половины пациентов сонливость и катаплексия существенно влияют на повседневную жизнь, включая учебу, работу, брак или общественную жизнь.
Пациенты, страдающие нарколепсией, могут извлечь пользу из регулярного и адекватного графика сна, запланированных дневных снов, отказа от лекарственных средств, вызывающих дневную сонливость или бессонницу, а также путем посещения групп психосоциальной поддержки. Скрининг на депрессию, гипертонию и
Подробности ➞
Многие расстройства сна, такие как апноэ во время сна, периодические движения ног, могут сосуществовать с нарколепсией, тем самым способствуя появлению ее симптомов. Данные расстройства должны быть устранены, прежде чем начинать лечение препаратами, специфическими при нарколепсии.
Большинство лекарственных средств, предлагаемых для терапии нарколепсии, направлены против либо дневной сонливости, либо катаплексии. Поэтому многим пациентам, у которых в наличии оба симптоматических состояния, для лечения нарколепсии требуется более одного препарата.
Несмотря на оптимальную терапию, немногие пациенты с нарколепсией когда-либо чувствуют себя полностью бодрыми. Целью лечения нарколепсии является повышение бдительности до такой степени, чтобы производительность и безопасность находились на адекватном уровне при таких важных видах деятельности, как учеба или работа. После достижения оптимума в терапии нарколепсии следует оценивать тяжесть остаточной сонливости по Эпвортской шкале сонливости (ESS) или тестом поддержания бодрствования (MWT). Пациенты, испытывающие постоянную и непреходящую сонливость, должны получить консультацию на предмет избегания потенциальных опасных видов деятельности вроде вождения автомобиля.
Для пациентов с тяжелой степенью сонливости, требующей медикаментозного лечения, предложено организовать первичную терапию при помощи модафинила (modafinil), а не метилфенидата (methylphenidate) или амфетамина (amphetamine). Связано это с тем, что модафинил был изучен среди пациентов с нарколепсией, пройдя проверку в рандомизированных, плацебо-контролируемых клинических испытаниях, притом что он модафинил, скорее всего, оказывает меньше побочных эффектов, чем традиционные стимуляторы. Однако нет никаких оснований подозревать, что терапевтическая эффективность модафинила превосходит таковую у других стимуляторов.
Солриамфетол (solriamfetol), пероральный селективный ингибитор обратного захвата дофамина и норадреналина с эффектом пробуждения, характеризуется, похоже, побочными эффектами, аналогичными модафинилу, и может стать разумным вариантом первой линии терапии при сонливости. Питолисант (pitolisant), обратный агонист гистаминового H3-рецептора, располагает умеренной активностью в качестве стимулирующего бодрствование лекарственного средства.
При назначении препаратов, способствующих бодрствованию, необходимо предупреждать пациентов о риске возникновения серьезных сердечно-сосудистых и психиатрических побочных реакций. Рекомендовано отслеживать сердечный ритм и кровяное давление во время каждого посещения врача, поскольку гипертония часто встречается у людей с нарколепсией и усугубляется большинством лекарств.
В группе препаратов первой линии при катаплексии входят лекарственные средства, подавляющие фазу быстрого сна, такие как венлафаксин (venlafaxine), флуоксетин (fluoxetine), атомоксетин (atomoxetine). При этом предпочтителен венлафаксин в рецептуре замедленного высвобождения.
Если сонливость или катаплексия не улучшаются в ходе лечения вышеуказанными препаратами, можно применять оксибутират натрия (sodium oxybutirate), также известный как оксибат натрия (sodium oxybate).
У людей с нарколепсией риск автомобильных аварии повышен в три-четыре раза, притом что свыше трети пациентов попали в аварию как раз из-за сонливости. Врачам следует открыто обсуждать равно как указанный риски с пациентами, так и стратегии его уменьшения.
Нефармакологическое лечение нарколепсии
Существует ряд не относящихся к фармакотерапии вмешательств, которые могут помочь при нарколепсии. Впрочем, большинство пациентов всё же нуждаются в соответствующих лекарственных препаратах, уменьшающих сонливость и катаплексию.
Дрёма и гигиена сна
Пациентам с нарколепсией показаны один или два качественных 20-минутных периода дневного сна, хотя некоторые больные выигрывают исключительно от длительного сна. Так, если вздремнуть в обеденное время (13:00–14:00 часов), это зачастую приводит к улучшению бдительности и внимательности на срок от одного до трех часов, тем самым снижая потребность в стимуляторах во второй половине дня. Если это помогает, показан короткий дневной сон на работе или в учебном заведении.
Лишение (депривация) сна может усугубить симптомы нарколепсии, потому следует придерживаться регулярного и адекватного режима сна.
Лекарства и вещества
Необходимо избегать определенных лекарственных препаратов и веществ по причине их потенциальной способности ухудшать симптомы нарколепсии. Среди таковых, которые могут усилить дневную сонливость: бензодиазепины, опиаты, антипсихотики (нейролептики), алкоголь. Некоторые, такие как теофиллин (theophylline) или избыток кофеина, могут вызвать бессонницу, что в конечном итоге отразится повышенной дневной сонливостью.
Празосин (prazosin) и другие антагонисты альфа-1 адренергических рецепторов ухудшают симптомы катаплексии.
Психосоциальная поддержка
Пациенты, страдающие нарколепсией, на протяжении всей своей жизни сталкиваются с различными психосоциальными и связанными с работой проблемами, в результате чего испытывают трудности в выполнении экономических и социальных обязанностей. На них также ложится дополнительное бремя в преодолении ошибочных представлений о причинах и непроизвольном характере симптомов нарколепсии.
Распространенные ошибочные представления (даже среди тех, кто оказывает медицинскую помощь) заключаются в том, что приступы сна и катаплексия являются проявлениями слабой мотивации, отрицания или избегания. И потому пациентам обычно показано участие в группах поддержки, которые сосредоточены на навыках преодоления трудностей и выявлении ресурсов социальной помощи в решении административных и медицинских вопросов.
Поддержание здоровья при нарколепсии
Психиатрия
Пациенты с нарколепсией подвержены повышенному риску сопутствующих психических заболеваний — особенно депрессии и тревоги. По крайней мере раз в один-два года необходимо проходить скрининговое обследование на наличие депрессии.
Рекомендовано проходить опросник здоровья пациента (PHQ-2), поскольку он быстрый, достаточно чувствительный и лишен недостатков других инструментов скрининга вроде PHQ-9, которые содержат ненужные в данном случаи вопросы, относящиеся к утомляемости, проблемам с концентрацией внимания и фрагментированным сном. Опросник PHQ-2 содержит только два вопроса:
- в течение последнего месяца часто ли вас беспокоило чувство подавленности, депрессии или безнадежности?
- в течение последнего месяца часто ли вас беспокоило отсутствие интереса или удовольствия?
Сердечно-сосудистая система
Нарколепсия ассоциирована с более высокими, нежели ожидаемые, показателями артериального давления, что лишь частично связано с приемом стимуляторов и иных лекарственных препаратов.
В исследовании, которое проводило круглосуточное амбулаторное измерение артериального давления у взрослых (медиана возраста 32–41 лет) пациентов (n=160) с нарколепсией типа 1, распространенность гипертонической болезни составила 41% среди нелечившихся больных и 58% среди получавших стимулирующие лекарственные средства.
У пациентов с нарколепсией также более вероятны
Подробности ➞
Оценка и модификация факторов риска сердечно-сосудистой системы является важным компонентом поддержания здоровья у пациентов с нарколепсией.
Ожирение
" target="_blank" >ожирение широко распространены при нарколепсии. Это связано, вероятно, с дефицитом гипокретина, но остается неясным, связано ли
Подробности ➞
" target="_blank" >ожирение с низким базальным метаболизмом, сниженной физической активностью или повышенным потреблением калорий.
Значительный прирост массы тела на 9–14 кг не является чем-то необычным в период манифестации нарколепсии у детей, причем даже без очевидных изменений в потреблении пищи, вот почему снижение скорости метаболизма является вероятной причиной.
Рекомендованы регулярные физические упражнения, ограничение калорийности потребляемой пищи, консультация с диетологом для выработки конкретного плана по снижению веса.
Фармакологическое лечение нарколепсии
Для пациентов с нарколепсией, сопровождаемой повышенной дневной сонливостью, рекомендованы следующие лекарственные средства:
- модафинил (modafinil);
- в случае сохранения повышенной дневной сонливости применяется увеличенная доза модафинила или метилфенидата (methylphenidate);
- если повышенная дневная сонливость не отвечает на фармакотерапию, следует рассмотреть следующие варианты лекарственных средств:
- оксибутират натрия (sodium oxybutirate);
- модафинил и оксибутират натрия;
- метилфенидат и оксибутират натрия;
- питолизант (pitolisant);
- комбинация из метилфенидата и модафинила;
- армодафинил (armodafinil).
Для пациентов, страдающих нарколепсией с катаплексией, рекомендованы следующие лекарственные средства:
- оксибутират натрия;
- венлафаксин (venlafaxine);
- трициклические антидепрессанты, такие как кломипрамин (clomipramine), и другие антидепрессанты, включая флуоксетин (fluoxetine), циталопрам (citalopram) и сертралин (sertraline);
- селегилин (selegiline);
- питолизант.
Для пациентов, страдающих нарколепсией с нарушением сна ночью, рекомендованы следующие лекарственные средства:
- оксибутират натрия, в том числе в сочетании с модафинилом.
Для пациентов, страдающих нарколепсией с галлюцинациями и ночным параличом, рекомендованы следующие лекарственные средства:
Лечение нарколепсии у детей и подростков
Для пациентов младше 16 лет не существует одобренных лекарственных препаратов для терапии нарколепсии.
Пациентам следует придерживаться строгих графиков отхода ко сну и физической активности.
В педиатрической популяции распространено применение вне инструкции медикаментов для взрослых.
Синдром Коффина-Лоури, независимо описанный Коффином в 1966 году и Лоури в 1971 году, характеризуется значительной умственной отсталостью у мальчиков и различной или отсутствующей умственной отсталостью у гетерозиготных девочек . Фация характерна для подростков и взрослых. Кисти маленькие, пальцы слишком растянуты, часто маленькие и тонкие. У мальчиков наблюдается постоянная задержка роста. Микроцефалия является распространенным явлением. Иногда обнаруживается, что болезнь сердца способствует смерти от этого синдрома. Эпизоды внезапного падения без потери сознания, вызванного тактильным или слуховым раздражителем, возникают примерно в подростковом возрасте в 20% случаев. Позднее начало сколиоза - наиболее яркая черта этого заболевания.
Ген RPS6KA3 кодирует рибосомный протеин S6 киназу альфа 3, который действует на ATF4 , фактор транскрипции (белок, контролирующий экспрессию гена), необходимый для созревания остеобластов и синтеза коллагена I типа . Мутация гена RSK2 вызывает уменьшение количества зрелых остеобластов и уменьшение костной массы. Дефицит коллагена объясняет прогрессирующую деградацию позвонков, подвергающихся сильным ограничениям.
RSK2 также фосфорилирует CREB, фактор транскрипции.
Секвенирование позволяет найти мутацию в 40% случаев.
Резюме
Описание
Синдром Коффина-Лоури - это глубокая умственная отсталость, связанная с аномалиями
роста.
- Внутриутробный рост является нормальным, но послеродовой рост очень низкий, около третьего процентиля. Иногда появляется микроцефалия.
- Мелкий зуб, гиподонтия, ретрогнатия у молодых людей сменяется прогнатизмом.
Неврология и поведение
- Часто описывается как веселый и приятный, но это не постоянная черта. Эта патология также является причиной внезапных падений без потери сознания при воздействии зрительных и слуховых раздражителей. Эти проявления затрагивают около 20% пациентов.
Сердечно-сосудистые
- Сердечные аномалии встречаются у 15% пациентов.
- Кифоз прогрессирующего сколиоза поражает 1 из 2 пациентов.
Слух и зрение
- Нарушения слуха являются обычным явлением, и их следует искать. Нарушения зрения встречаются реже.
Диагностический
Диагностировать в раннем детстве непросто: черты лица, особенно черты лица, становятся очевидными только в подростковом возрасте.
Клинический
Наиболее важными клиническими признаками для постановки диагноза являются:
Дисморфизм лица.
О Со№п в 1966 г и В 1_о\л/гу в 1972 г независимо друг от друга описали нескольких больных со сходными клиническими проявлениями, включавшими низкий рост, умственную отсталость, антимонголоид- ный разрез глаз, луковицеобразный нос и конические пальцы В 1975 г 3 ТепИату, проанализировав собственные наблюдения и материалы, опубликованные в литературе, предложил назвать это заболевание синдромом Коффина-Лоури
Популяционная частота заболевания неизвестна
Гэнетические данные и патогенез.
Синдром Коффина-Лоури наследуется по Х-сцепленному доминантному типу с развернутыми клиническими проявлениями у мужчин и более стертыми - у женщин Ген данного синдрома локализован на коротком плече хромосомы X в локусе Хр22 2-Хр22 1
Патогенез заболевания остается неясным При помощи молекулярно-генетиче- ского анализа у больных с синдромом Коффина-Лоури удалось выявить делеции, нонсенс- и миссенс-мутации в гене проте инкиназы (Н5К2) Ген Н5К2 локализуется в регионе Хр22 3, в пределах которого располагается ген синдрома Коффина-Лоури [17] Установлено, что белок - продукт гена Н5К2 - активирует специфический белок- коактиватор генной экспрессии (СРЕВ) путем его фосфорилирования Существует предположение что белок СНЕВ вовлечен в процесс развития когнитивных функций и может влиять на формирование умственной отсталости при синдроме Коффина-Лоури [18] Ген Н5К2 также входит в семейство генов, участвующих в процессах регуляции клеточного цикла [19]
У больных с синдромом Коффина-Лоури выявляются изменения в структуре соединительной ткани при микроскопическом исследовании биоптатов кожи у некоторых пробандов определялось почти полное от сутствие эластических волокон, в биоптатах костной ткани - аномалии строения и незрелость хондроцитов Не исключено, что в патогенезе формирования скелетных аномалий участвуют обменные нарушения в коллагене и протеогликанах [20]
Полная картина заболевания обычно наблюдается у лиц мужского пола К основным клиническим признакам относятся низкий рост, ан- тимонголоидный разрез глаз, луковицеобразный нос, конусовидные пальцы, скелетные аномалии, умственная отсталость Черты лица очень специфичны квад ратный лоб, выступающие надбровные дуги, широко расставленные глаза с опущенными наружными углами, периорбиталь- ная полнота тканей, широкая спинка носа, вывернутые вперед ноздри, массивный подбородок, большой рот с толстыми выступающими губами и вывернутой нижней губой, большие оттопыренные ушные ра ковины Нередко обнаруживаются отсутствие боковых резцов, преждевременное выпадение молочных зубов Иногда встречаются утолщение уздечки, расщепление губы У некоторых больных описана компенсированная гидроцефалия
Нередко наблюдаются шейный лордоз, кифоз в грудном отделе, килевидная форма грудной клетки, короткая расщепленная грудина, тораколюмбальный сколиоз, укорочение длинных трубчатых костей нижних конечностей, плоскостопие Скелетные деформации с возрастом могут усугубляться У некоторых пациентов описано сужение позвоночного канала Больные отстают в росте
У отдельных больных описаны пороки развития внутренних органов односторонний гидронефроз, кишечные дивертикулы, митральная недостаточность, долевая эмфизема Могут наблюдаться сосудистые нарушения телеангиэктазии, варикозное расширение вен В некоторых случаях выявляются нарушения зрения
На основании 20-летнего наблюдения за шестью больными с синдромом Коффи- на-Лоури А (3 \Л/ Нип1ег суммировал клинически значимые осложнения, которые встречаются при этом синдроме преждевременная смерть, чаще от кардиоваскулярных осложнений, прогрессирующий кифосколиоз, который может ограничивать подвижность грудной клетки и вызывать сердеч но-сосуд истую недостаточ ность, стеноз позвоночного канала, приводящий к развитию радикуломиелопатии, катаплексия [23]
У женщин клинические проявления заболевания выражены стерто Характерно негрубое снижение интеллекта, имеются сведения о нормальном психическом развитии Описаны случаи развития депрессивного психоза Наиболее часто встречаются конусовидные изменения пальцев кистей, реже отмечаются выступающие надбровные дуги, утолщение носовой перегородки, толстые вывернутые губы Диагностика. Диагноз устанавливается на основании клинических проявлений и данных анализа родословной Возможно проведение ДНК-диагностики в высокоспециализированных и хорошо оснащенных лабораториях
При рентгенологическом исследовании отмечаются отставание костного возраста от паспортного, сужение межпозвоночных дисков, утолщение костей лицевого черепа, укорочение концевых фаланг пальцев, укорочение длинных трубчатых костей нижних конечностей, соха уа!да Дифференциальный диагноз синдрома Коффина-Лоури проводят с синдромом
Коффина-Сириса, Берьесона-Форсма- на-Лемана
Лечение носит симптоматический характер Применяют курсы ноотропных препаратов для улучшения психического развития больных, проводится ортопедическая коррекция нарушений осанки
Профилактика основана на данных ме- дико-генетического консультирования, исходя из Х-сцепленного доминантного типа наследования заболевания Особое внимание следует уделять обследованию женщин - возможных носительниц патологического гена, у которых наблюдаются минимальные клинические проявления
Синдром Коффина-Лоури — это редкая генетическая болезнь с Х-сцепленным механизмом передачи, которая характеризуется выраженной умственной отсталостью, множественными фенотипическими особенностями. Пациенты имеют специфические черты внешности: высокий лоб, гипертелоризм, крупный выступающий нос. Помимо интеллектуальных нарушений, наблюдаются задержка роста, тугоухость, кардиологические осложнения. Основу диагностики синдрома Коффина-Лоури составляет молекулярно-генетическое исследование. Лечение поддерживающее: специальная программа нейрореабилитации, коррекция костных искривлений, слухопротезирование при тугоухости.
МКБ-10
Общие сведения
Болезнь получила свое название в честь двух ученых, которые независимо друг от друга описали ее патогномоничные симптомы, — американского педиатра Грейнджа С. Коффина (1966 г.) и канадского генетика Роберта Б. Лоури (1971 г.). Молекулярно-генетические основы патологии были расшифрованы в 1996 году. Синдром Коффина-Лоури в полной клинической форме встречается только у больных мужского пола. Женщины, являющиеся носителями мутантного гена, могут иметь умеренные признаки синдрома. Заболевание регистрируется с частотой 1-2 случая на 100 000 живорожденных новорожденных.
Причины
Болезнь Коффина-Лоури связана с наличием мутации гена RPS6KA3 на Х-хромосоме. До 80% случаев патологии возникают спорадически в семьях без отягощенной наследственности, а оставшиеся 20% заболевших приходятся на Х-сцепленный рецессивный тип наследования. Клинические признаки проявляются преимущественно у мужчин. Факторы, способствующие спонтанным генетическим мутациям, пока не установлены.
Патогенез
В норме ген RPS6KA3 кодирует синтез белка киназы RSK2, необходимого для правильного протекания процессов пролиферации, дифференцировки, запрограммированной клеточной смерти. Вещество активизируется при помощи протеинов сигнального пути, начинает работать при поступлении гормонов роста, нейротрансмиттеров, полипептидных гормонов.
При дисфункции RPS6KA3 в организме наблюдается дефицит киназы, что становится причиной расстройств передачи по MARK-сигнальному пути. В результате этого ухудшается формирование долговременной памяти и других когнитивных функций, что обусловлено нарушениями синаптической передачи, а также возникают множественные патологии развития опорно-двигательного аппарата.
Симптомы
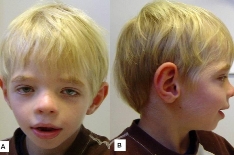
Наиболее характерные клинические проявления: многочисленные аномалии лицевого скелета. У детей с рождения высокий выступающий лоб, широко расставленные глаза с узкими глазными щелями. Переносица уплощенная, крылья носа крупные с вывернутыми ноздрями. Аномалии ротовой полости включают узкое высокое небо, изменения прикуса, срединную язычную борозду.
У страдающих синдромом Коффина-Лоури широкие кисти с короткими пальцами, которые резко сужаются к дистальным отделам. В 80% случаев возникает деформация грудной клетки, кифосколиоз. У мужчин отмечается серьезная задержка физического развития, средний рост взрослых составляет около 143 см (диапазон 115-158 см). Женщины-гетерозиготы зачастую имеют нормальную длину тела — более 50% пациенток находятся выше 10-го центиля.
С первых лет жизни у больных проявляется нарушение речевой функции, отставание в интеллектуальном развитии. Степень умственной отсталости широко варьирует, у разных пациентов уровень IQ может быть в диапазоне 15-60. При этом у ребенка частично сохранены коммуникативные навыки, характер обычно спокойный и дружелюбный. Тяжелые формы умственной отсталости, как правило, формируются при сопутствующей тугоухости.
Осложнения
До 20% больных страдают нарушениями координации движений, внезапными падениями (дроп-атаки), которые появляются в школьном возрасте. До 15% пациентов имеют сердечно-сосудистые осложнения (дисфункцию митрального клапана, кардиомиопатию). У 5% людей с синдромом Коффина-Лоури отмечаются эпилептические приступы. Гетерозиготные женщины часто имеют ожирение, психические проблемы: депрессии, шизофрению, психопатологический тип поведения.
Согласно литературным данным, около 13,5% мужчин погибают в 13-34 лет, остальные доживают до среднего возраста. Причинами летального исхода выступают негативные последствия синдрома Коффина-Лоури — миокардит, пневмония, дыхательная недостаточность. Также существует риск смерти от осложнений при проведении наркоза для ортопедической коррекции костных аномалий или выполнения стоматологических манипуляций.
Диагностика
Характерный фенотип в сочетании с нарушениями психомоторного развития позволяет заподозрить синдром Коффина-Лоури еще в младенчестве. Обследованием таких больных занимается врач-педиатр совместно с детским неврологом и генетиком. Чтобы подтвердить диагноз, необходима комплексная диагностика, включающая следующие методы:
- Генетическое тестирование. Секвенирование экзона для обнаружения типичной мутации RPS6KA3 — основной способ подтвердить наличие синдрома. Такая диагностика может проводиться другим членам семьи при подозрении на наследственный характер болезни.
- Рентгенография скелета. На снимках лицевого черепа обнаруживается утолщение костей, множественные деформации. Рентгенография позвоночника показывает кифосколиоз, узкие межпозвоночные промежутки. При исследовании конечностей выявляется укорочение трубчатых костей, широкие проксимальные и узкие дистальные фаланги пальцев.
- Консультация невролога. Для оценки степени когнитивных и речевых расстройств требуется комплексный неврологический осмотр, в дошкольном и школьном возрасте применяются специальные тесты и опросники. По показаниям назначается нейровизуализация (КТ голоного мозга, нейросонография, ЭЭГ).
- Пренатальная диагностика. В семье с отягощенной наследственностью целесообразно исследовать генотип плода еще во время беременности. Забор материала для молекулярно-генетического теста выполняется с помощью амниоцентеза, кордоцентеза во втором триместре.
Лечение синдрома Коффина-Лоури
Консервативная терапия
Этиотропное лечение не разработано, поэтому медицинская помощь сводится к устранению или уменьшению симптоматики, предотвращению осложнений синдрома Коффина-Лоури. Чтобы контролировать состояние здоровья, пациентам необходимо динамическое наблюдение у ряда специалистов (педиатр/терапевт, невролог, кардиолог, генетик). Соответственно клинической картине назначаются:
- Психотропные препараты. Противосудорожные и ингибиторы обратного захвата серотонина рекомендованы при часто повторяющихся дроп-атаках. Регулярный прием антиконвульсантов показан, если заболевание осложняется судорожными приступами.
- Ноотропы. Использование лекарств, улучшающих энергообмен и кровообращение в головном мозге, стимулирует становление интеллектуальных функций, улучшает краткосрочную и долговременную память.
- Слухопротезирование. Детям с кондуктивной тугоухостью установка слухового аппарата выполняется как можно раньше, в идеале — в первые 6 месяцев жизни ребенка, что способствует развитию речи, правильному формированию коммуникативных навыков.
Учитывая умственную отсталость большинства больных, при синдроме Коффина-Лоури требуется специальная нейрореабилитация. Занятия начинаются с раннего возраста при участии логопедов, сурдопедагогов, коррекционных педагогов. Дети не могут обучаться в обычной школе, им требуются занятия в инклюзивных классах либо специальные программы домашнего обучения.
Хирургическое лечение
Оперативная ортопедическая помощь оказывается при 3-4 степени сколиоза, сопровождающегося кардиореспираторными нарушениями. Больным показана хирургическая коррекция искривления позвоночника с применением механической системы стабилизации
Прогноз и профилактика
Женщины-носители поврежденного гена имеют нормальную продолжительность жизни, симптомы выражены слабо или вовсе отсутствуют, поэтому прогноз благоприятный. Менее оптимистичны прогнозы для пациентов-мужчин, которые зачастую умирают в молодом и среднем возрасте от соматических или неврологических осложнений. Профилактика синдрома Коффина-Лоури включает медико-генетическое консультирование пар с семейной предрасположенностью.
1. Coffin—Lowry syndrome/ Marques Pereira, P., Schneider, A., Pannetier, S.// European Journal of Human Genetics. — 2010. — №18.
2. Altered neurodevelopment associated with mutations of RSK2: a morphometric MRI study of Coffin-Lowry syndrome/ Kesler S. R., Simensen R. J., Voeller K., Abidi F// Neurogenetics. — 2007. — №8.
3. Coffin-Lowry syndrome: clinical and molecular features/ Hanauer, A., and I. D. Young// Journal of medical genetics. — 2002. — №10.
Читайте также: