
Синдром дабина джонсона реферат
Обновлено: 05.07.2024
Синдром Дабина-Джонсона – хроническое наследственное заболевание, проявляющееся непостоянной желтухой.
Что провоцирует / Причины Синдрома Дабина-Джонсона:
Синдром Дабина-Джонсона имеет аутосомно-рецессивный тип наследования. Генетический дефект заключается в появлении мутации в гене, кодирующем белок, который является ионным каналом, транспортером органических анионов (cMOAT).
В результате гепатобилиарный транспорт билирубина и органических анионов нарушается. В крови увеличивается содержание фракции конъюгированного билирубина, в моче - билирубинурия.
Конъюгационная желтуха (конъюгированная гипербилирубинемия) диагностируется, если содержание прямой фракции билирубина превышает 0,3 мг/дл (5,2 мкмоль/л), а показатель общего сывороточного билирубина более 2,0 мг/дл (34,2 мкмоль/л) или если содержание прямой фракции билирубина составляет более 15% от показателя общего сывороточного билирубина, который превышает 2,0 мг/дл.
Патогенез (что происходит?) во время Синдрома Дабина-Джонсона:
Основным звеном патогенеза является нарушение экскреции пигмента из гепатоцитов. приводящее к регургитации билирубина.
Симптомы Синдрома Дабина-Джонсона:
Синдром Дабина - Джонсона - очень редкое заболевание, встречающееся преимущественно у мужчин молодого возраста, в некоторых случаях - с рождения. Клиническая симптоматика более ярко выражена, чем при других формах гипербилирубинемии. Отмечают повышенную утомляемость, плохой аппетит, боли в правом подреберье вплоть до колик, поносы. Желтуха у наблюдавшихся нами больных была постоянной и сопровождалась нерезким кожным зудом. Диспепсические кризы возникали чаще, самочувствие всегда плохое. В двух наблюдениях диспепсическим расстройствам предшествовал 2-3-дневный продромальный период с легкой гиперемией зева, субфебрильной температурой. У некоторых больных заболевание десятилетиями протекает бессимптомно. Печень нормальных размеров или выступает на 1-2 см из-под края реберной дуги.
Печень больных не может адекватно экскротировать билирубин, бромсульфалсин и контрастные препараты для холецистографии. Вследствие этого выявляют отклонение от нормы содержания билирубина, бромсульфал ей новой пробы и активности щелочной фосфатазы, а также частое отсутствие тени желчного пузыря при холецистографии. При раздельном исследовднии фракции билирубина преобладает прямой билирубин. В соответствии с этим при синдроме Дабина-Джонсона наблюдается билирубинурия.
Диагностика Синдрома Дабина-Джонсона:
Физикальные методы обследования
- осмотр – желтушность кожных покровов и слизистых.
Лабораторные исследования
- общий анализ крови;
- общий анализ мочи;
- билирубин крови – повышение конъюгированного билирубина;
- билирубин мочи - повышен
- проба с фенобарбиталом – снижение уровня билирубина на фоне приема фенобарбитала;
- ферменты крови (АсНТ, АлАТ, ГГТП, ЩФ) – возможно умеренное повышение;
- бромсульфалеиновая проба – повышение уровня в сыворотке кривой выведения через 90 мин в сравнении с таковым через 45 мин;
- уровень общего копропорфирина в суточной моче – не изменен;
- уровень изомера копропорфирина типа I в суточной моче – увеличение. При наличии показаний:
- маркеры вирусов гепатита В, С, D – для исключения вирусных гепатитов.
Инструментальные и другие методы диагностики
- УЗИ органов брюшной полости (определение размеров и состояния паренхимы печени – обычно умеренно увеличены; размеры, форма, толщина стенок желчного пузыря и желчных протоков – не изменены, конкременты отсутствуют; размеры селезенки – нередко бывают увеличены);
- пероральная или внутривенная холецистография – запаздывание или полное отсутствиe контрастирования желчного пузыря и желчных протоков.
При наличии показаний:
- функционная биопсия печени – обнаружение в гепатоцитах печени характерного пигмента;
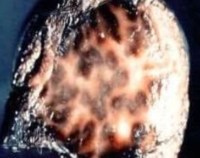
- диагностическая лапароскопия – характерное черное окрашивание печени.
Консультации специалистов
При наличии показаний:
- клинический генетик – с целью верификации диагноза.
Дифференциальная диагностика
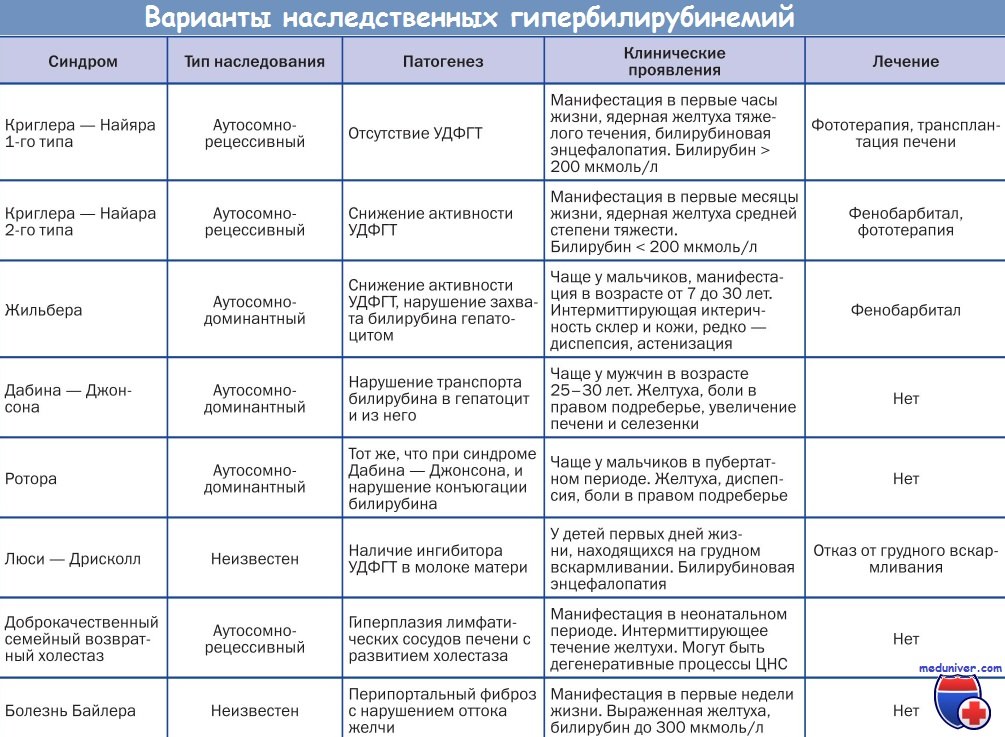
Проводится с другими гипербилирубинемиями (синдром Криглера-Найяра, Жильбера, Ротора), вирусным гепатитом, хроническим гепатитом проявляющимся холестатическим синдромом, механической желтухой, первичным билиарным циррозoм. В диагностике помогают особенности бромсульфанеиновой пробы, малоизменены другие функциональные пробы, чaсто семейный характер и начало болезни в детстве или юношеском возрасте. При необходимости - лапароскопия, пункционная биопсия.
Лечение Синдрома Дабина-Джонсона:
- Стремление избежать провоцирующих факторов (инфекции, физические и психические нагрузки, употребление алкоголя и гепатотоксичных лекарств)
- Противопоказана инсоляция
- Диета с ограничением тугоплавких жиров и продуктов содержащих консерванты. Витамины группы В.
- Рекомендуются желчегонные средства.
- Санация хронических очагов инфекции и лечение имеющейся патологии желчевыводящих путей.
Критерии эффективности лечения
Уменьшение интенсивности или устранение желтухи. Нормализация (достоверное уменьшение) уровня билирубина в крови. Продолжительность лечения – в течение всей жизни.
Профилактика Синдрома Дабина-Джонсона:
Родители, имеющие детей, страдающих этим синдромом, должны проконсультироваться у генетика перед планированием очередной беременности. Аналогичным образом следует поступать, если у родственников семейной пары, собирающейся иметь детей, диагностирован.
К каким докторам следует обращаться если у Вас Синдром Дабина-Джонсона:
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Синдрома Дабина-Джонсона, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору .
Синдром Дабина-Джонсона — это хроническое наследственное заболевание, которое характеризуется нарушением выделения билирубина из гепатоцитов в желчь. Основное клиническое проявление — интермиттирующая желтуха. Для болезни также характерны диспепсические расстройства, снижение аппетита и ухудшение общего самочувствия. Диагностика синдрома Дабина-Джонсона включает биохимические анализы крови и мочи, бромсульфалеиновую пробу, инструментальные методики (УЗИ, лапароскопию, биопсию печени). Лечение предполагает коррекцию образа жизни, назначение щадящей диеты. При необходимости используют желчегонные препараты и другие лекарственные средства.
МКБ-10
Общие сведения
Синдром имеет несколько синонимов — энзимопатическая желтуха, генетически обусловленный пигментный гепатоз. Нозологическая форма названа в честь двух американских ученых — И.Н. Дабина и Ф.Б. Джонсона, описавших синдром в 1954 году. Это редкое заболевание, в 70% проявляющееся в молодом возрасте. В основном синдром Дабина-Джонсона наблюдается у жителей Среднего Востока. Наибольшая частота встречаемости среди иранских евреев — 1 случай на 1300 населения. У 60% больных патология сопровождается снижением активности факторов коагуляции и кровотечениями. Заболеваемость не зависит от пола.
Причины
Синдром имеет генетическую природу, характеризуется аутосомно-рецессивным путем наследования. Среди родственников болезнь может проявляться как у мужчин, так и у женщин, повторяется через 1-2 поколения. Наследственный дефект представлен мутацией в нуклеотидной последовательности, обеспечивающей кодирование белка MRP2. Этот протеин отвечает за экскрецию (выделение) конъюгированного билирубина и органических анионов в желчные ходы.
Патогенез
Вследствие дисфункции АТФ-зависимой транспортной системы канальцев желчь не проходит в желчные капилляры, из-за чего билирубин накапливается в печеночных тканях. Длительно существующий синдром сопровождается обратными биохимическими реакциями: избыточное количество прямого билирубина освобождается от глюкуроновой кислоты. Полученный непрямой билирубин также проникает в кровоток, обуславливает токсическое действие на нервную систему.
Симптомы
Обычно манифестация синдрома Дабина-Джонсона происходит в 20-30-летнем возрасте, хотя первые характерные признаки появляются уже у подростков. Крайне редко заболевание выявляется у детей. У женщин с бессимптомным течением энзимопатической желтухи клинический дебют болезни могут спровоцировать наступление беременности или прием контрацептивных средств.
Основный симптом заболевания — желтуха, не сопровождаемая кожным зудом. Сначала возникает желтушность склер и слизистых оболочек, затем кожа также приобретает желтушный оттенок. Желтуху могут усиливать изнурительные физнагрузки, стрессовые ситуации, интеркуррентные инфекции. Как правило, желтушные периоды сменяются безжелтушными, хотя у ряда пациентов иктеричность кожи сохраняется постоянно.
Синдром периодически обостряется. Пациент жалуется на сильные боли справа в подреберье, реже — в околопупочной области. Иногда боль отдает в правое плечо или лопатку. Болевой синдром может быть настолько интенсивным, что напоминает печеночную колику. Одновременно с болью беспокоят тошнота, горечь во рту. Изредка бывает рвота, которая не приносит облегчения. Общая интоксикация проявляется повышенной утомляемостью, сонливостью, снижением аппетита.
Осложнения
Синдром Дабина-Джонсона отличается доброкачественным течением и при правильном лечении не вызывает неприятных последствий. Частое осложнение при длительно протекающем заболевании — воспалительные процессы в желчном пузыре и протоках. Воспаление может переходить и на печеночную паренхиму, вызывая холестатический гепатит. При наличии этих патологий желтуха у больных синдромом Дабина-Джонсона становится постоянной.
При присоединении бактериальной инфекции может возникать эмпиема желчного пузыря и гнойный холангит, которые при неблагоприятных условиях трансформируются в ограниченный перитонит. У пожилых людей наследственный пигментный синдром провоцирует появление фиброзных изменений в печеночных дольках. В результате снижается функциональная активность печени. Вследствие нарушения синтеза факторов свертывания крови возрастает риск кровотечений.
Диагностика
При физикальном обследовании пациентов врач-гастроэнтеролог или гепатолог пальпирует выступающий на несколько сантиметров из-под реберной дуги край печени. Синдром дифференцируют с болезнью Ротора, гепатитами, раковыми метастазами в печень. Симптомы поражения желчного пузыря (Мюсси, Ортнера, Кера) отрицательные. Для подтверждения заболевания используют следующие лабораторные и инструментальные исследования:
- Анализы крови. Основным диагностическим критерием синдрома является повышение уровня общего билирубина более 85 мкмоль/л, при этом массовая доля прямого билирубина превышает 15%. При выполнении коагулограммы у 60% пациентов выявляют снижение активности протромбина и уменьшение протромбинового времени.
- Анализ мочи. В моче обнаруживают повышенный уровень билирубина. Для дифференциальной диагностики с близким по клинической симптоматике синдромом Ротора оценивают соотношение копропорфиринов I и III типа в моче. При гепатозе Дабина-Джонсона количество копропорфирина 1 типа составляет 80%, а 3 типа — 20%.
- Бромсульфалеиновая проба. Анализ наиболее чувствителен для оценки печеночных функций. При синдроме Дабина-Джонсона спустя 45 минут концентрация в крови специального красителя, который предварительно ввели внутривенно, составляет более 6%. Такая проба считается положительной и указывает на снижение поглотительно-выделительной функции печени.
- УЗИ органов брюшной полости. При сонографии синдром проявляется увеличением печени на 1-2 см, у пациентов среднего и пожилого возраста нередко обнаруживают очаги фиброза. Размеры и контуры желчного пузыря не изменены, конкременты не визуализируются. Характерно одновременное увеличение селезенки.
- Инвазивные методы. В сомнительных ситуациях показана диагностическая лапароскопия, которая позволяет врачу осмотреть наружную поверхность печени и выявить характерные коричневые пятна. Чтобы подтвердить диагноз, назначается чрескожная биопсия печеночной ткани с последующим осмотром биоптатов под электронным микроскопом.
Лечение синдрома Дабина-Джонсона
Этиопатогенетическое лечение заболевания не разработано. Главная роль в предупреждении ухудшений состояния больных принадлежит немедикаментозным мероприятиям. Чтобы предотвратить обострения синдрома Дабина-Джонсона, пациентам рекомендуют по возможности избегать физического переутомления и стрессов. Основные направления лечения, применяемые в современной гастроэнтерологии для улучшения качества жизни человека:
- Диета. В рационе питания ограничивают потребление тугоплавких жиров и продуктов, содержащих консерванты. Полностью исключают употребление спиртных напитков. Показана витаминизированная диета с достаточной калорийностью.
- Препараты с желчегонным эффектом. Назначаются синтетические или растительные лекарственные средства, которые либо увеличивают концентрацию желчных кислот, либо повышают содержание водного компонента желчи. Специалисты отдают предпочтение мягким растительным препаратам.
- Санация очагов инфекции. Проводится выявление и лечение самых распространенных хронических источников инфекции — кариеса зубов и хронического тонзиллита. При обнаружении сопутствующей патологии желчевыводящих путей подбирается соответствующая патогенетическая терапия.
Прогноз и профилактика
Наличие болезни Дабина-Джонсона не влияет на продолжительность жизни и работоспособность, поэтому прогноз благоприятный. Синдром не прогрессирует. При соблюдении врачебных рекомендаций пациенты чувствуют себя хорошо. Больным необходимо воздерживаться от алкоголя и приема контрацептивов. Специфическая профилактика не разработана. Семьям с синдромом Дабина-Джонсона перед планированием беременности необходимо проконсультироваться с врачом-генетиком.
2. Клинические особенности течение синдрома Дабина-Джонсона у детей/ В.С. Березенко, М.Б. Диба, Ю.П. Резников// Современная педиатрия. — 2018.
4. Пигментные гепатозы: клинические особенности, пункционная биопсия, электронная микроскопия, диагноз, прогноз/ С.Д. Подымова// Практическая гастроэнтерология. — 2018.
Синдром Дубина-Джонсона представляет собой еще один тип семейной желтухи. Это доброкачественное состояние, хотя печень ребенка с данным синдромом имеет сниженную способность секретировать некоторые органические анионы и контрастное вещество при проведении холецистографии.
Причина возникновения этого синдрома — мутация гена MDR2. У пациентов (обычно не в младенческом, а в более старшем возрасте) развивается конъюгированная гипербилирубинемия, не связанная с холестазом. Данный синдром — еще один пример дефекта канальцевого переносчика гепатоцита.
Результаты традиционных анализов, назначаемых для исследования функции печени, в норме, за исключением повышенного уровня общего билирубина (обычно менее 15 мг/дл) с преобладанием конъюгированного билирубина. Согласно данным морфологических исследований, печень имеет черный цвет из-за накопления меланиноподобного пигмента в лизосомах.
Синдром Ротора
Синдром Ротора похож на синдром Дубина-Джонсона, за исключением того, что при синдроме Ротора не отмечается пигментации гепатоцитов и секреция печенью контрастного вещества при проведении холецистографии — нормальная. Кроме того, первоначальный дефект у младенцев с синдромом Ротора заключается в дефиците внутриклеточных белков, связывающих анионы и служащих переносчиками их в клетку.
В качестве белка-переносчика может выступать глютатион-5-трансфераза. Дефицит этого внутриклеточного белка приводит к тому, что конъюгаты билирубина снова попадают в кровь вместо того, чтобы быть экскретированными после прохождения через канальцевую мембрану. При обоих описываемых синдромах отмечается повышение экскреции копропорфирина с мочой. Распознавание синдрома Ротора и синдрома Дубина-Джонсона представляется важным в плане предупреждения проведения ненужных дополнительных диагностических исследований.
У умерших детей с гепатоцеллюлярным холестазом при гистологическом исследовании печени было выявлено значительное количество аномалии, включая выраженное перипортальное воспаление и фиброз, а также диффузную трансформацию гигантских клеток. Примерно у 30% младенцев с гепатоцеллюлярным холестазом развивается прогрессирующая печеночная недостаточность, еще у 10%, переживших первые месяцы болезни, позднее выявляется хроническая патология, в т.ч. цирроз. Остальные 60% детей полностью выздоравливают.
В целом исходы для пациентов с этим заболеванием в настоящее время значительно улучшились благодаря успехам трансплантации печени.
Видео этиология, патогенез желтухи (повышения билирубина)
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Что такое синдром Жильбера? Причины возникновения, диагностику и методы лечения разберем в статье доктора Васильева Романа Владимировича, гастроэнтеролога со стажем в 15 лет.
Над статьей доктора Васильева Романа Владимировича работали литературный редактор Маргарита Тихонова , научный редактор Сергей Федосов

Определение болезни. Причины заболевания
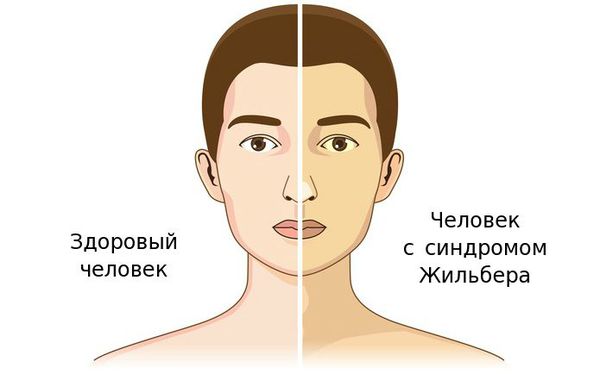
Синдром Жильбера — это генетический пигментный гепатоз с аутосомно-доминантным типом наследования, протекающий с повышением уровня неконъюгированного (свободного) билирубина, чаще проявляющееся в период полового созревания и характеризующийся доброкачественным течением [1] .
Краткое содержание статьи — в видео:
Синонимы названия болезни: простая семейная холемия, конституциональная или идиопатическая неконъюгированная гипербилирубинемия, негемолитическая семейная желтуха.
По распространённости данное заболевание встречается не менее, чем у 5 % населения, в соотношении мужчин и женщин — 4:1. Впервые заболевание описал французский терапевт Августин Жильбер в 1901 году.
Чаще синдром Жильбера проявляется в период полового созревания и характеризуется доброкачественным течением. Основным проявлением этого синдрома является желтуха.
К провоцирующим факторам проявления синдрома можно отнести:
- голодание или переедание;
- жирную пищу;
- некоторые лекарственные средства;
- алкоголь;
- инфекции (грипп, ОРЗ, вирусный гепатит);
- физические и психические перегрузки;
- травмы и оперативные вмешательства.
Причина заболевания — генетический дефект фермента УДФГТ1*1, который возникает в результате его мутации. В связи с этим дефектом функциональная активность данного фермента снижается, а внутриклеточный транспорт билирубина в клетках печени к месту соединения свободного (несвязанного) билирубина с глюкуроновой кислотой нарушается. Это и приводит к увеличению свободного билирубина.
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы синдрома Жильбера
Некоторые специалисты трактуют синдром Жильбера не как болезнь, а как физиологическую особенность организма.
До периода полового созревания данный синдром может протекать бессимптомно. Позже (после 11 лет) возникает характерная триада признаков:
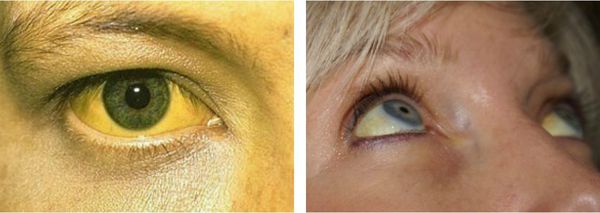
- желтуха различной степени выраженности;
- ксантелазмы век (жёлтые папулы);
- периодичность появления симптомов [1] .
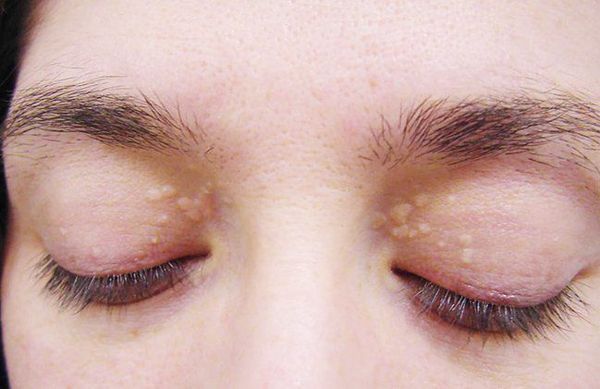
Желтуха чаще всего проявляется иктеричностью (желтушностью) склер, матовой желтушностью кожных покровов (особенно лица), иногда частичным поражением стоп, ладоней, подмышечных впадин и носогубного треугольника.
Заболевание нередко сочетается с генерализованной дисплазией (неправильным развитием) соединительной ткани.
Усиление желтухи может наблюдаться после перенесения инфекций, эмоциональной и физической нагрузки, приёма ряда лекарственных препаратов (в частности, антибиотиков), голодания и рвоты.
Клиническими проявлениями заболевания общего характера могут быть:
- слабость;
- недомогание;
- подавленность;
- плохой сон;
- снижение концентрации внимания.
В отношении ЖКТ синдром Жильбера проявляется снижением аппетита, изменением привкуса во рту (горечь, металлический привкус), реже возникает отрыжка, тяжесть в области правого подреберья, иногда наблюдается боль ноющего характера и плохая переносимость лекарственных препаратов.
При ухудшении течения синдрома Жильбера и существенном повышении токсичной (свободной) фракции билирубина может появляться скрытый гемолиз, усиливая при этом гипербилирубинемию и добавляя в клиническую картину системный зуд.
Патогенез синдрома Жильбера
В норме свободный билирубин появляется в крови преимущественно (в 80-85 % случаев) при разрушении эритроцитов, в частности комплекса ГЕМ, входящего в структуру гемоглобина. Это происходит в клетках макрофагической системы, особенно активно в селезёнке и купферовских клетках печени. Остальная часть билирубина образуется из разрушения других гемсодержащих белков (к примеру, цитохрома P-450).
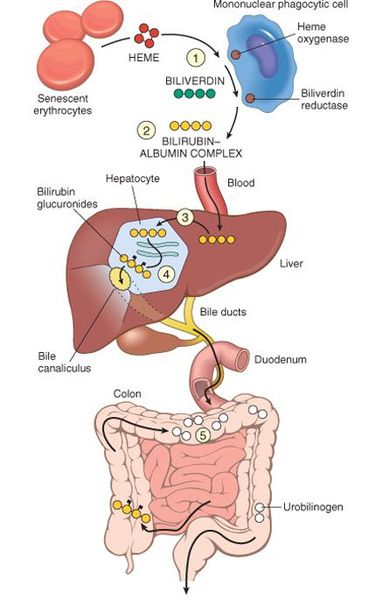
У взрослого человека в сутки образуется приблизительно от 200 мг до 350 мг свободного билирубина. Такой билирубин слаборастворим в воде, но при этом хорошо растворяется в жирах, поэтому он может взаимодействовать с фосфолипидами ("жирами") клеточных мембран, особенно головного мозга, чем можно объяснить его высокую токсичность, в частности токсичное влияние на нервную систему.
Первично после разрушения комплекса ГЕМ в плазме билирубин появляется в неконъюгированной (свободной или несвязанной) форме и транспортируется с кровью при помощи белков альбуминов. Свободный билирубин не может проникнуть через почечный барьер за счёт сцепления с белком альбумином, поэтому сохраняется в крови.
В печени несвязанный билирубин переходит на поверхность гепатоцитов. С целью снижения токсичности и выведения в клетках печени свободного билирубина при помощи фермента УДФГТ1*1 он связывается с глюкуроновой кислотой и превращается в конъюгированный (прямой или связанный) билирубин. Конъюгированный билирубин хорошо растворим в воде, он является менее токсичным для организма и в дальнейшем легко выводится через кишечник с желчью.
При синдроме Жильбера связывание свободного билирубина с глюкуроновой кислотой снижается до 30% от нормы, тогда как концентрация прямого билирубина в желчи увеличивается.
В основе синдрома Жильбера лежит генетический дефект — наличие на промонторном участке A(TA)6TAA гена, кодирующего фермент УДФГТ1*1, дополнительного динуклеотида ТА. Это становится причиной образования дефектного участка А(ТА)7ТАА. Удлинение промонторной последовательности нарушает связывание фактора транскрипции IID, в связи с чем уменьшается количество и качество синтезируемого фермента УДФГТ1, который участвует в процессе связывания свободного билирубина с глюкуроновой кислотой, преобразуя токсичный свободный билирубин в нетоксичный связанный.
Вторым механизмом развития синдрома Жильбера является нарушение захвата билирубина микросомами сосудистого полюса клетки печени и его транспорта глутатион-S-трансферазой, которая доставляет свободный билирубин к микросомам клеток печени.
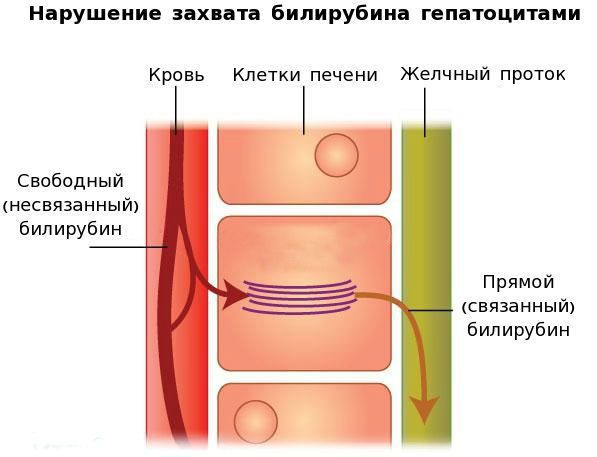
В конечном итоге вышеперечисленные патологические процессы приводят к увеличению содержания свободного (несвязанного) билирубина в плазме, что обуславливает клинические проявления заболевания [6] .
Классификация и стадии развития синдрома Жильбера
Общепринятой классификации синдрома Жильбера не существует, однако условно можно разделить генотипы синдрома по полиморфизму.
Читайте также: