
Синдром беквита видемана реферат
Обновлено: 28.06.2024
Некоторые дети рождаются с генетическими заболеваниями, которые могут привести к онкозаболеваниям.
Считается, что большинство видов онкозаболеваний у детей не являются наследственными (не передаются из поколения в поколение). Исследование, проведенное специалистами научно-клинического центра St. Jude Children's Research Hospital и Вашингтонского университета в рамках проекта генома детского рака, показало, что около 8,5% случаев рака у детей обусловлены исходным генетическим заболеванием.
Наличие такого заболевания не означает, что у человека обязательно возникнет рак. Оно свидетельствует лишь о повышенном риске из-за состава генов.
Такими генетическими заболеваниями являются:
Атаксия-телеангиэктазия
Атаксия-телеангиэктазия является редким наследственным заболеванием, которое поражает нервную систему, иммунную систему и другие системы организма. У детей с этим заболеванием наблюдаются прогрессирующие трудности с координацией движений, часто возникают сложности при ходьбе, проблемы с равновесием и координацией рук, непроизвольные подергивания, мышечные судороги и нарушения функции нервной системы. У них также повышен риск развития онкологических заболеваний, особенно лейкоза и лимфомы. Люди с атаксией-телеангиэктазией очень чувствительны к воздействию излучения, включая медицинское рентгеновское излучение. Узнайте больше об атаксии-телеангиэктазии.
Синдром Беквита-Видемана и изолированная гемигипертрофия
Синдром Беквита-Видемана — это генетическое нарушение, которое вызывает чрезмерный рост частей тела (гипертрофия). Он может быть ограничен одной областью тела или затрагивать несколько частей тела. Патология с одной стороны называется гемигипертрофией. У детей с синдромом Беквита-Видемана повышен риск развития рака, особенно одной формы рака почки, называемой опухолью Вильмса, и формы рака печени, называемой гепатобластомой. Опухоли развиваются примерно у 10% людей с этим заболеванием и почти всегда появляются в детстве. Узнайте больше о синдроме Беквита-Видемана.
Комплекс Карни
Комплекс Карни — патология, характеризующаяся повышенным риском развития нескольких видов опухолей, в том числе нераковых (доброкачественных) опухолей в области сердца, известных как миксомы сердца. Миксомы также могут появляться на коже и внутренних органах. У людей с комплексом Карни также может развиться рак органов эндокринной (гормонпродуцирующей) системы, таких как надпочечники, щитовидная железа, яички, яичники и гипофиз. Часто эта патология также влечет за собой изменения в окраске кожи (пигментация). Признаки и симптомы этого заболевания обычно проявляются в подростковом или раннем взрослом возрасте. Узнайте больше о комплексе Карни.
Синдром конституционального дефицита репарации неспаренных оснований ДНК
Синдром конституционального дефицита репарации неспаренных оснований ДНК — это редкое заболевание, которое повышает вероятность развития у ребенка нескольких различных форм рака, включая опухоли головного мозга, лейкоз, лимфомы, аномальные образования в кишечнике, желудочно-кишечном тракте и женских репродуктивных органах (матке и яичниках). У детей также могут появиться кожные изменения: пятна цвета кофе с молоком (плоские участки коричневого цвета на коже) или более светлые участки кожи по сравнению с общим тоном кожи. У ребенка с синдромом конституционального дефицита репарации неспаренных оснований ДНК может развиться несколько видов опухолей одновременно или в течение жизни. Узнайте больше о синдроме конституционального дефицита репарации неспаренных оснований ДНК.
DICER1-синдром
DICER1-синдром — это наследственное заболевание, которое повышает риск образования различных раковых и нераковых (доброкачественных) опухолей. Наиболее часто встречаются определенные виды опухолей в легких , почках , яичниках и щитовидной железе . У людей с таким синдромом может развиться один или несколько видов опухолей. У членов одной семьи могут развиваться разные виды. Узнайте больше о DICER1-синдроме.
Семейный аденоматозный полипоз (САП)
Семейный аденоматозный полипоз (САП) — это заболевание, которое в основном влияет на пищеварительную систему. У людей с семейным аденоматозным полипозом обычно наблюдается аномальный рост ткани (полипы) в толстой и тонкой кишке. Некоторые полипы могут стать злокачественными, если их не удалить. Для людей с семейным аденоматозным полипозом повышается риск развития рака толстой кишки и других видов рака пищеварительной системы в молодом возрасте. Кроме того, для них повышается риск развития других опухолей или физических проблем. Узнайте больше о семейном аденоматозном полипозе.
Наследственная нейробластома
Нейробластома — это вид рака, который возникает из незрелых нервных клеток, называемых нейробластами. Он в основном поражает детей в возрасте младше 5 лет и редко встречается у взрослых. Нейробластома возникает, когда нейробласты неконтролируемо растут, образуя опухоль. В большинстве случаев опухоль зарождается в нервной ткани надпочечников . Иногда нейробластомы возникают в нервных клетках брюшной полости , груди, шеи или таза . Узнайте больше о наследственной нейробластоме.
Наследственный синдром параганглиом/феохромоцитом
Наследственный синдром параганглиом/ феохромоцитом — это заболевание, при котором опухоли развиваются в структурах, называемых параганглиями . Узнайте больше о наследственном синдроме параганглиом/феохромоцитом.
Наследственная ретинобластома
Ретинобластома — это злокачественная опухоль глаза, чаще всего диагностируется у детей младше 5 лет. Рак развивается в области глаза, которая называется сетчаткой . Наследственная форма ретинобластомы обычно поражает оба глаза, но в некоторых случаях она может развиваться только в одном глазу. У людей с наследственной ретинобластомой повышается риск возникновения опухолей в других частях тела: на коже, в костях, мягких тканях и головном мозге. Узнайте больше о наследственной ретинобластоме.
Синдром Ли-Фраумени (СЛФ)
Синдром Ли-Фраумени (СЛФ) — это редкое заболевание, которое повышает вероятность развития одного или нескольких видов рака у больного в течение жизни. Этот синдром обычно наследуется от члена семьи.
Множественная эндокринная неоплазия 1-го типа
Множественная эндокринная неоплазия 1-го типа (МЭН 1) — это генетическое заболевание, которое повышает риск развития раковых и нераковых опухолей. Большинство из этих опухолей развиваются в органах эндокринной системы . Узнайте больше о множественной эндокринной неоплазии 1-го типа.
Множественная эндокринная неоплазия 2-го типа
Множественная эндокринная неоплазия 2-го типа — это генетическое заболевание, которое повышает риск развития рака в органах эндокринной системы . Для людей со множественной эндокринной неоплазией 2-го типа повышается риск развития медуллярного рака щитовидной железы (МРЩЗ) и феохромоцитом — опухолей надпочечников . У людей с этим заболеванием могут также развиться другие виды аномальных образований в тканях или железах эндокринной системы. Узнайте больше о множественной эндокринной неоплазии 2-го типа.
Нейрофиброматоз 1-го типа
Нейрофиброматоз 1-го типа — это наследственное заболевание, которое может поражать многие участки тела. Это может быть кожа, глаза, кости, кровеносные сосуды и нервная система. У людей с нейрофиброматозом 1-го типа также повышается риск развития некоторых видов раковых и нераковых опухолей. Это такие заболевания, как нейрофибромы , опухоли центральной нервной системы, феохромоцитомы , лейкозы и гастроинтестинальные стромальные опухоли (ГИСО). Тяжесть заболевания и поражаемые участки тела могут варьироваться от человека к человеку. Узнайте больше о нейрофиброматозе 1-го типа.
Нейрофиброматоз 2-го типа
Нейрофиброматоз 2-го типа — это генетическое заболевание, которое в основном поражает нервную систему. У людей с нейрофиброматозом 2-го типа повышается риск развития опухолей в нервах. Наиболее распространенный вид опухоли у людей с нейрофиброматозом 2-го типа — вестибулярная шваннома (другое название — акустическая невринома). У пациентов с нейрофиброматозом 2-го типа вестибулярные шванномы обычно развиваются в обоих слуховых нервах в среднем в возрасте 18–24 лет. Почти у всех пациентов с нейрофиброматозом 2-го типа к 30 годам появятся двусторонние вестибулярные шванномы. Узнайте больше о нейрофиброматозе 2-го типа.
Синдром злокачественного базальноклеточного невуса (синдром Горлина)
Синдром злокачественного базальноклеточного невуса — это заболевание, при котором повышается риск развития определенных видов раковых и нераковых опухолей. Опухоли могут развиваться в коже, челюсти, сердце, яичниках и головном мозге. Люди с синдромом злокачественного базальноклеточного невуса могут также иметь физические отличия, такие как большой размер головы (иногда в результате избытка жидкости в головном мозге), мелкие белые шишки на лице, лишние пальцы рук и ног, расщелина губы и неба, а также умственные трудности и проблемы с обучением. Узнайте больше о злокачественном базальноклеточном невусе.
Синдром Нунан
Для людей с синдромом Нунан несколько повышается риск развития некоторых видов рака и заболеваний крови. Это миелопролиферативные нарушения , ювенильный миеломоноцитарный лейкоз (ЮММЛ), нейробластома и эмбриональная рабдомиосаркома. У людей с синдромом Нунан немного повышается риск развития определенных нераковых опухолей, таких как гигантоклеточные поражения и зернистоклеточные опухоли . Узнайте больше о синдроме Нунан.
Синдром Пейтца-Егерса
Синдром Пейтца-Егерса — это генетическое заболевание, которое повышает риск развития у человека некоторых видов рака. К ним относятся рак желудка, пищевода, кишечника, поджелудочной железы, толстой кишки, прямой кишки, молочной железы, матки, шейки матки, легких, яичников и яичек. У людей с синдромом Пейтца-Егерса также повышается риск развития доброкачественных опухолей (полипов) в пищеварительной системе. Злокачественные и доброкачественные опухоли могут также появляться в почках, легких, желчном пузыре , носовых ходах, мочевом пузыре или мочеточниках . У людей с синдромом Пейтца-Егерса на некоторых участках кожи часто могут быть маленькие темные пятна, похожие на веснушки. Узнайте больше о синдроме Пейтца-Егерса.
PTEN-ассоциированный синдром гамартомных опухолей
PTEN-ассоциированный синдром гамартомных опухолей — это генетическое заболевание, при котором в различных частях тела возникают доброкачественные опухоли (гамартомы). Помимо гамартом, у пациентов могут быть и другие физические отклонения, в том числе увеличенный размер головы, аномальные кожные наросты и трудности в обучении. У людей с PTEN-ассоциированным синдромом гамартомных опухолей повышается риск развития рака молочной железы, щитовидной железы, почек, матки, колоректального рака и рака кожи. Узнайте подробнее о PTEN-ассоциированном синдроме гамартомных опухолей.
Синдром предрасположенности к рабдоидным опухолям
У людей с синдромом предрасположенности к рабдоидным опухолям повышается риск их появления. Эти быстрорастущие опухоли часто встречаются в головном мозге, спинном мозге и почках. Также могут развиваться опухоли мягких тканей, легких, кожи и сердца. Эти опухоли чаще всего встречаются у детей. Для людей с синдромом предрасположенности к рабдоидным опухолям также может повышаться риск развития нераковых опухолей, растущих в нервах — шванном . Эти опухоли чаще встречаются у взрослых. Узнайте больше о синдроме предрасположенности к рабдоидным опухолям.
Комплекс туберозного склероза
Комплекс туберозного склероза (КТС) — это генетическое нарушение, которое может приводить к появлению нераковых опухолей в коже, головном мозге, почках и других органах. Иногда эти опухоли вызывают значительные проблемы со здоровьем. Комплекс туберозного склероза также вызывает проблемы с развитием. Признаки и симптомы заболевания варьируются. Почти все больные имеют кожные аномалии, в том числе пятна необычайно светлой кожи, участки выступающей и утолщенной кожи и наросты под ногтями. Опухоли на лице — лицевые ангиофибромы — также часто появляются в детстве. Узнайте больше о синдроме туберозного склероза.
Синдром фон Гиппеля-Линдау
Синдром фон Гиппеля-Линдау — это редкое заболевание, при котором у человека повышается вероятность развития некоторых нераковых и раковых опухолей. У людей с синдромом фон Гиппеля-Линдау могут развиться опухоли центральной нервной системы и сетчатки ( гемангиобластомы ), опухоли внутреннего уха ( опухоли эндолимфатического мешка ), кисты или рак почек, кисты или рак поджелудочной железы, опухоли надпочечников ( феохромоцитомы ) и опухоли половых путей ( папиллярные цистаденомы ). Узнайте больше о синдроме фон Гиппеля-Линдау.
WT1-ассоциированные синдромы
WT1-ассоциированные синдромы предрасположенности к опухоли Вильмса — это заболевания, поражающие почки. У людей с этими заболеваниями повышается риск развития раковой опухоли почек — опухоли Вильмса. У людей с WT1-ассоциированными синдромами могут быть другие медицинские проблемы. К ним могут относиться проблемы с репродуктивными органами и глазами, а иногда — проблемы с поведением или развитием. Узнайте больше о WT1-ассоциированных синдромах предрасположенности к опухоли Вильмса.
Х-сцепленный лимфопролиферативный синдром
Х-сцепленный лимфопролиферативный синдром (ХЛПС) — это очень редкая патология, при которой иммунная система не работает надлежащим образом. У людей с ХЛПС чаще развивается тяжелая реакция, которая называется молниеносным инфекционным мононуклеозом (МИМ). При МИМ активируется слишком много иммунных клеток, которые накапливаются в различных частях тела, включая печень, селезенку, костный мозг и головной мозг. Эту реакцию иногда также называют гемофагоцитарным лимфогистиоцитозом (ГЛГ). Люди с ГЛГ почти всегда мужчины. Заболевание ежегодно диагностируется менее чем у одного из миллиона мальчиков или юношей. Узнайте больше об Х-сцепленном лимфопролиферативном синдроме (ГЛГ).
Пигментная ксеродерма
Пигментная ксеродерма (ПК) — врожденное патологическое состояние, значительно повышающее чувствительность к ультрафиолетовым (УФ) солнечным лучам. Это заболевание в основном затрагивает глаза и участки кожи, подвергающиеся воздействию солнца. У некоторых больных также возникают проблемы с нервной системой. Люди с пигментной ксеродермой имеют значительно повышенный риск развития рака кожи. Без защиты от солнца около половины детей с этой патологией заболевают первым раком кожи к 10 годам. У большинства людей с пигментной ксеродермой рак кожи возникает в течение жизни несколько раз. Чаще всего поражаются лицо, губы и веки. Рак также может развиться на коже головы, в глазах и на кончике языка. Исследования показывают, что у людей с пигментной ксеродермой также может быть повышен риск развития других видов онкологических заболеваний, таких как опухоли головного мозга. Кроме того, курящие больные значительно чаще страдают от рака легких. Узнайте больше о пигментной ксеродерме.
—
Дата изменения: декабрь 2018 г.
Генетические консультанты и сведения о риске
Генетический консультант — это медицинский работник, специализирующийся на медицинской генетике и консультировании в этой области; он может помочь семьям получить информацию.
Синдром Беквита-Видемана — это сложная мультигенная патология, которая проявляется множественными врожденными пороками развития. Заболевание возникает при различных генетических аномалиях 11 хромосомы в сочетании с эпигенетическими нарушениями. Триада признаков болезни Беквита-Видемана включает макроглоссию, омфалоцеле, макросомию. Также наблюдаются разнообразные соматические патологии. Диагностика представлена генетическим исследованием, визуализацией внутренних органов методами УЗИ, МРТ, КТ. План лечения и реабилитации подбирается индивидуально, в соответствии с тяжестью синдрома, числом и характером клинических симптомов.
МКБ-10
Q87.3 Синдромы врожденных аномалий, проявляющихся избыточным ростом [гигантизмом] на ранних этапах развития
Общие сведения
Причины
Патология возникает, когда сочетаются генетические причины (различные мутации ДНК) и эпигенетические факторы (метилирование ДНК, биохимические изменения гистонов). До 85% случаев синдрома проявляются спорадически, а оставшиеся 15% наследуются от родителей по аутосомно-доминантному типу с неполной пенетрантностью. Нарушения структуры 11-й хромосомы у страдающих болезнью Беквита-Видемана подразделяются на следующие группы:
- Нарушение метилирования IC2. Самый частый вид эпигенетической мутации, который выявляется у 50% пациентов. В большинстве случаев такие расстройства появляются у детей без отягощенной наследственности.
- Отцовская однородительская дисомия. При этом варианте синдрома ребенок наследует обе хромосомы из конкретной пары от одного родителя, что чревато развитием тяжелых врожденных пороков.
- Мутация CDKN1C. Такая генная аномалия имеет 50% вероятность передачи от больной матери ребенку, спорадические мутации встречаются крайне редко.
- Гиперметилирование IC1 (H19). Этот тип эпигенетических нарушений составляет до 5% всех случаев синдрома Беквита-Видемана. Он бывает без делеции (спорадические варианты) или с делецией (наследуется от матери).
- Аномалии локуса 11p15. Менее 1% пациентов составляют люди с цитогенетически видимой дупликацией хромосомы или ее транслокацией/инверсией. Такие мутации в основном наследуются от отца и матери.
Патогенез
В основе всех возникающих дефектов лежат молекулярные нарушения в генах короткого плеча 11 хромосомы. Здесь располагаются центры импринтинга, которые отвечают за правильную экспрессию генов, полученных от матери с отцом. У здоровых людей эти гены контролируют рост органов и тканей, угнетают избыточную пролиферацию клеток. Соответственно при их мутации у плода происходят множественные органные нарушения, формируются врожденные пороки скелета.
Симптомы
Среди нарушений внутренних органов превалируют медуллярная дисплазия почек, цитомегалия фетальной коры надпочечников, висцеромегалия — увеличение размеров печени, селезенки, поджелудочной железы. Реже встречаются пороки развития сердца, врожденная кардиомиопатия, кардиомегалия. После рождения у младенца обычно выявляется тяжелая неонатальная гипогликемия с характерным симптомокомплексом.
Осложнения
Наличие эпигенетических мутаций создает предпосылки для развития эмбриональных опухолей. Самые распространенные типы онкопатологии у больных с синдромом Беквита-Видемана — гепатобластома, нефробластома, рабдомиокарцинома, аденокарцинома. Из-за множественных врожденных аномалий, которые усугубляются полиорганными нарушениями, у пациентов сохраняется высокая вероятность летального исхода. В детском возрасте умирает до 20% больных.
Диагностика
- Молекулярно-генетическое тестирование. При проведении генетической диагностики учитывается возможность гетерогенности синдрома, поэтому исследования делают сразу на несколько типичных мутаций, эпигенетических аномалий. Все дети с проявлениями синдрома Беквита-Видемана также проходят обязательное кариотипирование.
- Инструментальная визуализация. Специфическим признаком синдрома считается висцеромегалия, для обнаружения которой выполняется УЗИ, КТ или МРТ органов брюшной полости. Для диагностики кардиомегалии показана эхокардиография, а сопутствующие нарушения работы сердца регистрируются на ЭКГ.
- Неврологический осмотр. Обследование новорожденного у детского невролога необходимо для исключения сопутствующих поражений ЦНС, родовых травм на фоне макросомии. В будущем требуется оценка когнитивных способностей, которые могли пострадать вследствие неонатальной гипогликемии.
Лечение синдрома Беквита-Видемана
Специфическая терапия болезни еще не разработана. Лечение проводится мультидисциплинарной командой врачей и направлено на устранение или уменьшение отдельных проявлений синдрома Беквита-Видемана. Согласно клиническим протоколам, в план оказания медицинской помощи при этой генетической патологии включаются:
- Коррекция гипогликемии. Это самый важный момент лечения в неонатальном периоде, поскольку нормальный уровень гликемии необходим для функционирования головного мозга. Коррекция выполняется 10% растворами глюкозы, при необходимости вводятся глюкокортикоиды.
- Химиолучевая терапия. Такое лечение назначается при онкологических заболеваниях, которые зачастую возникают у детей раннего возраста в результате наследственной предрасположенности.
- Пластическая коррекция. Участие ортопедов и пластических хирургов требуется для устранения деформаций и аномальной длины конечностей, восстановления правильных пропорций тела, повышения функциональных возможностей опорно-двигательного аппарата.
С учетом типичных признаков и возможных осложнений синдрома разрабатывается комплексная программа диспансерного наблюдения пациента, для чего привлекают ортопедов, эндокринологов, неврологов, кардиологов. По показаниям применяются реабилитационные мероприятия с участием дефектологов, логопедов, специалистов ЛФК и массажистов.
Прогноз и профилактика
Синдром Беквита-Видемана сопряжен с высоким риском инвалидизирующих осложнений, а у 1/5 пациентов наблюдается летальный исход. Прогноз сомнительный, он определяется степенью выраженности фенотипических изменений, числом врожденных соматических патологий. Поскольку болезнь принадлежит к категории мультигенных с преимущественно спорадическим развитием, эффективные меры профилактики пока не разработаны.
2. Тактика амбулаторного наблюдения за пациентом с синдромом Беквита-Видемана/ Э.А. Каширина, А.А. Рубцова, Н.М. Югай, О.Б. Карабанова// Медицинский совет. — 2017. — №19.
3. Макросомия плода: современное состояние проблемы/ Р.С. Геворкян, А.Н. Рымашевский, А.Е. Волков, В.В. Маркина// Современные проблемы науки и образования. — 2016. — №6.
4. Рождение ребенка с синдромом Видемана-Беквита у пациентки после применения программы ЭКО/ИКСИ (клинический случай)/ Т.А. Назаренко, Н.А. Зыряева// Проблемы репродукции. — 2014. — №3.
Синдром Видемана-Беквита (синдром омфалоцеле, макроглоссии, гигантизма, EMG-синдром) впервые описан в 1963 году. У новорожденных была отмечена гипогликемия в сочетании с соматическими изменениями.
Что провоцирует / Причины Синдрома Видемана-Беквита:
Тип наследования заболевания – аутосомно-доминантный. В некоторых случаях выявляют структурные перестройки 11 хромосомы.
Симптомы Синдрома Видемана-Беквита:
Макроглоссия выявляется в любом возрасте ребенка, часто отмечается с рождения. Язык может не помещаться во рту, за счет чего рот ребенка открыт, а лицо напоминает таковое у больного с гипотиреозом. Увеличенный язык затрудняет сосание и даже дыхание новорожденного, у более старших детей отмечают дизартрические расстройства.
Гипогликемия у новорожденного манифестирует уже на первые-третьи сутки. Развивающиеся за счет этого коматозные состояния могут повлечь за собой смерть ребенка на первом году жизни или тяжелое органическое поражение мозга, сопровождающееся умственной отсталостью. Развитие гипогликемических состояний связано с гиперплазией островковых клеток поджелудочной железы, приводящей к гиперинсулинемии. С начинающейся еще внутриутробно гиперпродукцией инсулина, обладающего анаболическим действием, связывают как макроглоссию, макросомию, висцеромегалию, так и предрасположенность к развитию опухолей паренхиматозных органов. Явления гипергликемии самопроизвольно убывают в течение первых месяцев жизни больного.
Склонность к увеличению массы тела отмечается уже при рождении, она обычно превышает 4000 г, а длина – 52 см. Внутриутробная висцеромегалия, по-видимому, является причиной образования различных грыж, в том числе характерного для синдрома омфалоцеле (пуповинной грыжи). Пуповинная грыжа диагностируется у новорожденного и может быть различной по величине, иногда достигая размеров детской головки.
Макросомия с увеличением мышечной ткани и подкожного жирового слоя отмечается с рождения или развивается постнатально. Постнатальный гигантизм относится к менее постоянным признакам, иногда проявляется увеличением одной половины тела.
К каким докторам следует обращаться если у Вас Синдром Видемана-Беквита:
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Синдрома Видемана-Беквита, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору .
Кафедра акушерства, гинекологии, перинатологии и репродуктологии факультета последипломного профессионального обучения врачей Московской медицинской академии им. И.М. Сеченова; Научный центр акушерства, гинекологии и перинатологии им. акад. В.И. Кулакова
Центр лечения бесплодия "ЭКО", Москва
Рождение ребенка с синдромом Видемана-Беквита у пациентки после применения программы ЭКО/ИКСИ (клинический случай)
Кафедра акушерства, гинекологии, перинатологии и репродуктологии факультета последипломного профессионального обучения врачей Московской медицинской академии им. И.М. Сеченова; Научный центр акушерства, гинекологии и перинатологии им. акад. В.И. Кулакова



Представлено клиническое наблюдение - рождение ребенка с синдромом Видемана-Беквита у пациентки после применения программы ЭКО/ИКСИ.
Кафедра акушерства, гинекологии, перинатологии и репродуктологии факультета последипломного профессионального обучения врачей Московской медицинской академии им. И.М. Сеченова; Научный центр акушерства, гинекологии и перинатологии им. акад. В.И. Кулакова
Центр лечения бесплодия "ЭКО", Москва
В основе импринтинга лежат различия в экспрессии генов, обусловленные метилированием ДНК. У человека предполагают наличие 300-500 импринтированных генов. В основном они кодируют ростовые факторы, одни экспрессируют с отцовской, другие - с материнской хромосомой 6. Клетки каждого человека содержат отпечатки (характер метилирования) от обоих родителей. При гаметогенезе эти отпечатки стираются и устанавливается характер метилирования, присущий женскому или мужскому организму. При оплодотворении эмбрион получает по одному набору от каждого родителя с соответствующим характером экспрессии. В случае попадания в процессе оплодотворения двух гомологичных хромосом от одного родителя это состояние называется однородительской дисомией (ОРД) [5, 6].
Нарушения метилирования возможны при гаметогенезе, в процессе оплодотворения и преимплантационного развития. Нарушения метилирования могут происходить на разных уровнях организации генома (геномный, хромосомный, генный), вызывая соответствующие отклонения в развитии или болезни импринтинга [4, 6, 7]. Наиболее распространены и изучены у человека синдромы Прадера-Вили, Ангельмана, Видемана-Беквита, Сильвера-Рассела и некоторые другие.
Синдром Видемана-Беквита, или EMG-синдром по первым буквам характерной триады - омфалоцеле, макроглоссия, гигантизм, впервые описан патологом J. Beckwith (США) в 1963 г. и педиатром H.-R. Wiedemann (Германия) в 1964 г. Синдром встречается с частотой 1 случай на 10-12 тыс. новорожденных, при этом 85% - спорадические случаи и только 15% - наследственные (аутосомно-доминантный тип наследования с неполной пенетрантностью, передача по материнской линии) [8].
Критерии диагностики в настоящее время четко не установлены, но диагноз ставится при наличии 3 больших и 1 малого диагностического критерия. Из больших критериев наиболее характерны макросомия, макроглоссия, омфалоцеле, аномалии почек, эмбриональные опухоли. Из малых критериев характерны многоводие, неонатальная гипогликемия. Дифференциальный диагноз проводится со следующими синдромами: синдромом Симпсона-Голаби-Бехмеля (макросомия, висцеромегалия, макроглоссия, аномалии почек; Х-сцепленный рецессивный тип наследования), синдромом Перлмана, синдромом Костелло, синдромом Сотоса, синдромом Марото-Лами (мукополисахаридоз IV типа) [8].
Молекулярной основой синдрома Видемана-Беквита являются нарушения в генах короткого плеча хромосомы 11. Участок на коротком плече 11-й хромосомы включает две группы импринтированных генов - домен 1 (гены ИПФР2, Н19 и ЦИ1) и домен 2 (гены CDKN1C, KCNQ1 и ЦИ2) (ЦИ - центр импринтинга) [8].
Этиология синдрома сложна и включает 5 групп нарушений. Нарушения метилирования 1 и 2 ЦИ (потеря метилирования ЦИ2 материнского аллеля CDKN1C - 50% и гиперметилирование ЦИ1 материнского аллеля ИПФР2 и Н19 - 5%), а также отцовская ОРД по 11-й хромосоме - 20% - встречаются наиболее часто. Нередки мутации гена CDKN1C материнского аллеля: спорадические - 5%, наследственные - 40%. Также встречаются дупликации, инверсии, транслокации в этих доменах - 1% и субмикроскопические нарушения - частота не определена. В 20% случаев характер нарушений не известен [8].
Учитывая возможность нарушений метилирования в процессе гаметогенеза, оплодотворения и преимплантационного развития, встает закономерный вопрос о влиянии вспомогательных репродуктивных технологий (ВРТ) на частоту возникновения болезней импринтинга. По данным литературы 12, многие авторы отмечают повышенный риск болезней импринтинга после применения ВРТ. Некоторые авторы [15, 16] не отмечают повышения риска. Другие связывают болезни импринтинга у потомства с исходными нарушениями у родителей при бесплодии [17]. Также имеются отдельные клинические наблюдения [18, 19].
Представляем клиническое наблюдение.
Пациентка И., 1980 года рождения, рост 176 см, масса 62 кг. Наследственность не отягощена. Из соматического анамнеза обращает на себя внимание наличие пролапса митрального клапана, гипотонии и с 12 лет брадикардии. В детстве состояла на учете по поводу хронического пиелонефрита, с учета снята: аппендэктомия в 16 лет; страдает хроническим колитом. Является носителем вируса простого герпеса (ВПГ1), периодически отмечает обострения лабиального герпеса. Менархе в 12 лет, менструальный цикл регулярный. Половая жизнь с 18 лет. Беременностей не было. Брак первый с 2008 г., муж - 1978 года рождения, здоров. Гинекологический анамнез отягощен оперативными вмешательствами: 2 раза было экстренное чревосечение по поводу разрыва эндометриоидных кист яичников (в 2004 и 2008 г.). В 2006 г. была проведена лапароскопия, разделение спаек в малом тазу и гистерорезектоскопия, рассечение неполной внутриматочной перегородки. В 2012 г. - гистероскопия, раздельное диагностическое выскабливание перед проведением повторного цикла ЭКО.
Женщина обратилась по поводу бесплодия в 2008 г. Диагноз: первичное бесплодие; наружный генитальный эндометриоз IV степени; спаечный процесс в малом тазу; седловидная матка; хронический эндометрит.
Проведено 3 цикла ЭКО/ИКСИ по длинному протоколу (агонисты ГнРГ + чМГ + рФСГ), при пункциях получено по 3 ооцита, проведен перенос 2 эмбрионов на 3-и сутки, беременность наступила в 3-м цикле, одноплодная.
Беременность протекала с угрозой прерывания в I триместре, с 23 нед выявлено многоводие, с 27 нед - двусторонний уретерогидронефроз у плода по данным УЗИ. В 34 нед беременности отмечено многоводие крайней степени выраженности по данным УЗИ, нарушение кровотока у плода по данным допплеровского исследования, произведено кесарево сечение в экстренном порядке, родился живой недоношенный мальчик массой тела 2850 г, длиной 46 см, оценка по шкале Апгар 3-7 баллов. Мама выписана на 10-е сутки, послеоперационый период протекал без осложнений.
Мальчик родился в октябре 2009 г. Диагноз основной: множественные врожденные пороки развития; порок центральной нервной системы (незрелость структур головного мозга); макросомия; макроглоссия; неонатальный сахарный диабет; правосторонний пузырно-мочеточниковый рефлюкс III-IV степени; дефект межжелудочковой перегородки. Осложнения: церебральная ишемия II степени; геморрагический синдром (легочное и желудочное кровотечение). Сопутствующий: тяжелая асфиксия при рождении; внутриутробная пневмония; анемия новорожденных; недоношенность 34 нед.
В родильном зале ребенку произведена интубация трахеи, переведен на искусственную вентиляцию легких. Ребенок наблюдался в течение 1 мес жизни в отделении интенсивной терапии новорожденных. Отмечались плотные распространенные отеки туловища, синдром угнетения ЦНС (снижен мышечный тонус и угнетены рефлексы), в последующем на фоне терапии отмечена положительная динамика. По поводу гипергликемии с рождения получал инсулинотерапию (актропид внутривенно капельно), был взят генетический анализ на неонатальный сахарный диабет. Был установлен уретральный катетер, затем наложена пункционная цистостома, после чего признаки дилатации чашечно-лоханочной системы и обоих мочеточников по данным УЗИ купированы.
По данным магнитно-резонансной томографии (МРТ) выявлено нарушение формирования структур головного мозга: картина наружной гидроцефалии, гипоплазии височных долей, нарушения процесса формирования борозд головного мозга, задержки миелинизации. При консультации генетиком на основании наличия синдромальной формы макросомии и множественных врожденных пороков развития поставлен диагноз: синдром Симпсона-Голаби-Бехмеля. Взят цитогенетический анализ. Кариотип 46XY, нормальный мужской.
В возрасте 1 мес ребенок переведен в ДГКБ №1. На первом году жизни произведены операции по поводу паховой грыжи, глоссомегалии. В 7 мес жизни на консультации генетика установлен диагноз: синдром Видемана-Беквита (макросомия, макроглоссия, задержка психомоторного развития - гипоплазия височных долей по данным МРТ). Ребенок наблюдался у невролога, челюстно-лицевого хирурга. В 2 года на консультации генетика поставлен диагноз: синдром Видемана-Беквита (макроглоссия, мегауретер с гидронефротической трансформацией. Аномалия головного мозга на МР-томограмме. Задержка темпов психомоторного развития. Гипертензионно-гидроцефальный синдром). Ребенок получал лечение у нефролога, невролога, проходил скрининг 4 раза в год по поводу возможных эмбриональных опухолей (УЗИ, альфа-фетопротеин крови).
Пациентка повторно обратилась с целью достижения беременности в августе 2012 г. Естественно возник вопрос о возможностях профилактики и диагностики синдрома Видемана-Беквита при повторной беременности у женщины. Пациентка была проконсультирована генетиками из Медико-генетического центра РАМН. К сожалению, в настоящее время точная лабораторная диагностика болезней импринтинга четко не разработана, поэтому молекулярно-генетическое обследование ребенка и супружеской пары не проводилось. При медико-генетическом консультировании синдром Видемана-Беквита у ребенка был расценен как спорадический, поэтому риск при повторной беременности был расценен как низкий.
Учитывая упомянутые молекулярные особенности, лежащие в основе болезней импринтинга, проведение преимплантационной генетической диагностики (ПГД) также не представлялось возможным.
Проведен 4-й цикл ЭКО - по длинному протоколу (агонисты ГнРГ, чМГ, рФСГ), при трансвагинальной пункции получено 2 ооцита, 27.09.12 перенесено 2 эмбриона 3-го дня развития, наступила вторая беременность (последняя менструация 09.09.12).
В I триместре у женщины был впервые выявлен первичный субклинический гипотиреоз, в течение всей беременности пациентка принимала эутирокс 75-50 мкг/сут. В остальном беременность протекала без особенностей.
По результатам 1-го и 2-го скринингов и УЗИ отклонений от нормы не отмечено. При медико-генетическом консультировании показаний для проведения инвазивной пренатальной диагностики не выявлено. Кроме того, в отношении болезней импринтинга возможности пренатальной диагностики также четко не установлены.
Согласно информации, полученной из источников в Интернете, методы лабораторной диагностики синдрома Видемана-Беквита, имеющиеся на современном этапе развития медицины, находятся на стадии разработки и лишь частично внедрены в практику, в основном в США, странах Европы. Методы лабораторной диагностики зависят от генетической этиологии и включают следующие способы: 1) анализ метилирования с помощью метилчувствительной множественной полимеразной цепной реакции (МЧ мПЦР; MS-MLPA); МЧ ПЦР; 2) анализ однородительской дисомии с помощью исследования однонуклеотидного полиморфизма, МЧ мПЦР (MS-MLPA) и ПЦР; 3) определение мутаций с помощью анализа последовательностей; 4) определение дупликаций, инверсий и транслокаций с помощью цитогенетического анализа и FISH и, возможно, современного метода сравнительной геномной гибридизации; 5) выявление субмикроскопических нарушений с помощью анализа микроделеций и микродупликаций [7, 8, 20, 21].
Тактика медико-генетического консультирования также зависит от генетической причины синдрома Видемана-Беквита, последняя определяет и степень риска для сибсов пробанда [8].
У 85% больных с синдромом Видемана-Беквита наследственность не отягощена и кариотип нормальный. При этом высокий риск (50%) существует при наличии мутаций гена CDKN1C и микроделеций, микродупликаций; в остальных случаях риск низкий. В этих случаях проводят определение мутаций у пробанда и родителей, а также в семье.
При невыясненной причине (мозаицизм при ОРД) риск эмпирически низкий.
У 10-15% больных наследственность отягощена и нормальный кариотип.
При выявлении мутации CDKN1C у пробанда (40%): при наличии мутации у одного из родителей риск составляет 50%, при отсутствии мутации у родителей риск низкий. Но возможен мозаицизм. При отсутствии мутации CDKN1C у пробанда (60%) риск для сибсов составляет до 50%.
Пренатальная диагностика проводится следующим образом [8].
В случае наличия больного ребенка в семье: при выявлении мутаций проводят амниоцентез и анализ ДНК. Теоретически возможен анализ метилирования в амниоцитах. При всех беременностях риска: в 16 нед при наличии омфалоцеле определяют альфа-фетопротеин крови, проводят УЗИ в 19-20 нед и 25-32 нед беременности для выявления пороков развития и макросомии.
При неотягощенной наследственности: в случае выявления изолированного омфалоцеле при УЗИ возможен амниоцентез, анализ метилирования и мутации гена СDKN1С, дупликации, инверсии, транслокации. Проводят динамическое УЗИ во время беременности для выявления пороков развития и макросомии.
У новорожденного проводят мониторинг неонатальной гипогликемии. Диагноз подтверждают вышеуказанными способами [8].
Вопрос о преимплантационной диагностике не решен. Теоретически возможно применение метода сравнительной геномной гибридизации в случае отягощенной наследственности и наличия мутаций в семье.
В структуре наследственных заболеваний, сопровождающихся задержкой нервно-психического развития детей, высокий удельный вес составляют генетически детерминированные болезни, характеризующиеся высокими показателями физического развития, среди которых синдром Беквита-Видемана занимает одно из первых мест.
Синдром Беквита-Видемана получил свое название по имени немецкого педиатра Видемана (Мейетапп) и американского врача Беквита (ВескмуМ), впервые описавших это заболевание, соответственно, в 1963 и 1964 годах.
Частота патологии - 1 :10 000-1 :15 000 новорожденных [1].
Генетические данные и патогенез. Тип наследования синдрома неоднозначен и довольно сложен. В 15% семейных случаев характер передачи заболевания определяется как аутосомно-доминантный. У большинства больных (примерно 80%) патология возникает спорадически.
Синдром Беквита-Видемана обусловлен различными повреждениями кластера генов, расположенных в области короткого плеча хромосомы 11, в сегменте 15.5 [2].
Большую роль в нормальном функционировании генов этого локуса играет геномный импринтинг. Импринтинг - это различное действие генов в зависимости от их родительского (отцовского или материнского) происхождения. В настоящее время нарушение импринтинга считается основной причиной развития синдрома Беквита-Видемана.
У некоторых больных заболевание обусловлено хромосомной патологией - деле- цией, дупликацией или транслокацией с вовлечением локуса 15.5 короткого плеча хромосомы 11. На долю таких хромосомных аномалий приходится, примерно, 2% [3].
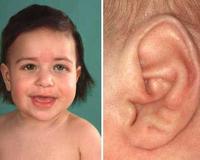
Клинические проявления. Заболевание формируется внутриутробно. Обращают на себя внимание высокие антропометрические параметры новорожденного: в среднем, длина тела составляет 52,6 см, масса - 4 кг. Высокие показатели физического развития сохраняются и в дальнейшем. Характерны также макро- глоссия и грыжа пупочного канатика (ом- фалоцеле).
Типичными признаками синдрома являются горизонтальные насечки на мочках ушей, микро- или макроцефалия, выступающий затылок, экзофтальм (вследствие относительной гипоплазии орбит), пупочная и паховая грыжи, клито- ромегалия, висцеромегалия (печени, почек, поджелудочной железы, иногда сердца). Нередко встречаются пороки развития внутренних органов: двурогая матка, диафрагмальная грыжа, добавочная селезенка, врожденные пороки сердца - чаще септальные дефекты, легких (неправильная дифференцировка легочной ткани на доли), кишечника (незавершенный поворот кишечника).
Психическое развитие больных варьирует: описаны пациенты как с нормальным интеллектом, так и с умственной отсталостью различной степени выраженности (рис. 7.2.1 на цветной вкладке).
Диагностика и дифференциальная диагностика. При обследовании новорожденных с синдромом Беквита-Видемана, как правило, выявляются полицитемия и гипогликемия, приводящие к развитию тяжелой неврологической симптоматики и, нередко, к летальному исходу.
У детей старше 7-10 лет часто диагностируют гиперлипидемию, гиперхолесте- ринемию, гипокальциемию [4].
Для детей с синдромом Беквита-Видемана характерно повышенное содержание в сыворотке крови соматомедина С, играющее, вероятно, ведущую роль в реализации высоких антропометрических параме-
трое больных [5] Высокий уровень сома- томедина С в сыворотке крови детей с синдромом Беквита-Видемана обусловлен избыточной продукцией гена инсули ноподобного фактора роста-2 (ген ЮР2), возникающей в результате потери геномного импринтинга (диаллельная экспрессия гена 1(3 Р2)
При синдроме Беквита-Видемана нередко регистрируются иммунодефицит- ные состояния Заболевание относится к группе риска развития злокачественных новообразований Одной из возможных причин формирования опухолей может быть снижение адаптивного ответа, выявляемое у больных с синдромом Беквита-Видемана [5] Суть его заключается в том, что предварительная обработка лимфоцитов малыми (неповреждающими) дозами мутагена с чувствительностью клеток к воздействию больших (повреждающих) доз этого же самого или других мутагенов Адаптивный ответ оценивается по процентному содержанию двуните- вой ДНК У здоровых детей адаптивный ответ, как правило, нормальный и равен 100% У лиц с синдромом Беквита-Виде- мана процент двунитевой ДНК после двойной обработки лимфоцитов у-лучам и в возрастающей дозировке составяет не выше 75%, что свидетельствует о полном отсутствии у больных адаптивного ответа по у-типу
Рентгенологические методы исследования позволяют выявить опережение костного возраста, расширение метафизов длинных трубчатых костей и кольцевидное сужение диафизов
Патоморфологические изменения при патолого-анатомическом анализе нередко обнаруживают гиперплазию клеток островков Лангерганса, нефрогенную бласто- му, резкое увеличение клеток и ядер в надпочечниках
Дифференциальная диагностика проводится с заболеваниями, которым свойственны высокие антропометрические параметры и снижение интеллекта К таким болезням относятся синдромы Сотоса, Симпсона-Голаби-Бехмеля, Вивера, Маршалла-Смита, аномалии половых хромосом (синдромы Клайнфельтера - 47, XXV и 47, ХУУ) и омфалоцеле
Лечение синдрома Беквита-Видемана носит симптоматический характер и направлено, в основном, на стимуляцию нер- вно-психического развития и иммунного статуса больных
С целью профилактики развития злокачественных новообразований необходимо оберегать больных от неблагоприятных воздействий внешней среды строго запрещены профессии, связанные с радиацией и химическими веществами, противопоказано проживание в районах с повышенным радиационным фоном и высокой инсоляцией Показан также строгий контроль за назначением цитостати- ческих препаратов и ионизирующего облучения
Читайте также: