
Реферат врожденные полипозные синдромы
Обновлено: 30.06.2024
Врожденные заболевания являются одной из распространенных причин младенческой и детской смертности, развития хронических болезней у детей и инвалидизации.
Быстрый переход
Врожденные заболевания могут быть обусловлены генетическими мутациями (передающимися по наследству или спонтанными), инфекциями матери во время беременности (цитомегаловирус, ветряная оспа, краснуха), воздействием лекарств и химических веществ, загрязняющих воздух, воду или пищу. Причины множества врожденных дефектов до сих пор неизвестны.
Генетическими или наследственными факторами обусловлены около 20 % врожденных заболеваний. К ним относятся нарушения, при которых мутация затрагивает один ген (серповидно-клеточная анемия); хромосомные нарушения, при которых хромосомы (или их части) отсутствуют (синдром Тернера) или имеют структурные изменения (увеличение количества хромосом или трисомия при синдроме Дауна); многофакторные нарушения, вызванные мутациями двух и более генов. Врожденные дефекты и нарушения развития могут вызывать делеции или дупликации отдельных генов (изменения митохондриальной ДНК). Примером такого заболевания является муковисцидоз, характеризующееся поражением экзокринных желез и жизненно-важных органов (легких и желудочно-кишечного тракта). Врожденные заболевания также ассоциированы со случайными (новыми) повреждениями генов, спонтанными (не наследующимися от родителей) мутациями (большинство случаев ахондроплазии).
По статистике, врожденные заболевания имеют 2–3 % младенцев. К возрасту 1 года их число увеличивается до 5 %, поскольку не все эти патологии диагностируются сразу после рождения.
Врожденные (наследственные) заболевания также классифицируют по типу наследования. При аутосомно-доминантном наследовании заболевание может передаваться от родителя к ребенку в 50 % случаев (мышечная дистрофия Дюшенна, хорея Гентингтона). При аутосомно-рецессивном наследовании генетическая аномалия передается ребенку только в том случае, если оба родителя наделены одним и тем же дефектным геном (муковисцидоз, серповидно-клеточная анемия). Здесь частота наследования составит 25 %, то есть в среднем у 1 ребенка из 4 детей этих родителей будет аутосомно-рецессивное заболевание.
Патологические состояния могут передаваться и при наследовании, сцепленном с полом (наследование гена, находящегося в половых хромосомах). Х-сцепленные рецессивные заболевания почти всегда ограничены мужским полом (гемофилия, дальтонизм, мышечная дистрофия Дюшенна). Х-сцепленные доминантные заболевания встречаются как у мальчиков, так и у девочек, однако у детей мужского пола имеют более тяжелое течение (Х-сцепленный гипофосфатемический рахит). Y-сцепленные заболевания встречаются довольно редко, поскольку Y-хромосома содержит всего несколько генов (ихтиоз).
Некоторые врожденные заболевания формально не относятся к генетическим, но имеют ту или иную выраженность наследования: наследуются факторы риска либо сам ген, но с низкой пенетрантностью (частотой проявления гена в признаках).
Гемофилия
Гемофилия — группа редких наследственных нарушений свертываемости крови, вызванных дефицитом необходимого белка (фактора свертывания крови).
Гемофилия A (классическая) встречается чаще (>80 % случаев) и связана с дефицитом VIII фактора свертывания крови, гемофилия B (болезнь Кристмаса) — реже (>10 % случаев), она обусловлена недостаточностью IX фактора свертывания крови. Гемофилия C встречается очень редко, обусловлена дефицитом XI фактора свертывания крови, чаще всего в классификации группы ее не упоминают.
Заболевание относится к X-сцепленным с полом, наследуется по рецессивному признаку по женской линии. Классическую гемофилию вызывают мутации гена F8, расположенного на X-хромосоме. Примерно в 70 % случаев заболевание наследуется по Х-сцепленному образцу, в остальных случаях оно возникает спонтанно (новая мутация), в дальнейшем спонтанное заболевание становится наследственным. Гемофилией A болеют практически исключительно мужчины, редкие случаи заболевания у женщин, носительниц дефектного гена, почти всегда характеризуются легким течением. Гемофилию B вызывают мутации гена F9, так же расположенного на X-хромосоме, она характерна для мужчин, женщины болеют очень редко. В некоторых случаях заболевание возникает спонтанно (приобретенная гемофилия A или B) и тоже связано с недостаточностью VIII или IX факторов свертывания крови. Приобретенная гемофилия является аутоиммунным заболеванием, при котором организм вырабатывает антитела, атакующие факторы свертывания крови (чаще всего VIII фактор). Примерно в половине случаев приобретенной гемофилии у пациента имеется связанное с ней основное состояние или заболевание (беременность, аутоиммунные заболевания, миелопролиферативные заболевания, воспалительные заболевания кишечника и др.), в остальных эпизодах причина остается невыясненной.
Гемофилия A затрагивает примерно 1 из 5 000 новорожденных мальчиков, гемофилия B — примерно 1 из 25 000. Около 60 % пациентов имеют тяжелую форму гемофилии, обычно им ставят диагноз при рождении или в течение первых 2 лет жизни.
Возраст манифестации гемофилии и тяжесть течения заболевания зависят от уровня активности факторов свертывания крови. Легкая форма характеризуется уровнем фактора свертывания крови (VIII или IX), превышающим 5 % от нормы, средняя — 1–5 %, тяжелая — ниже 1 %. У большинства пациентов, независимо от тяжести течения заболевания, эпизоды кровотечения чаще встречаются в раннем детском, детском и подростковом возрасте, чем в дальнейшей взрослой жизни.
При легкой и умеренной формах гемофилии длительные кровотечения могут возникнуть только в результате травмы, хирургического вмешательства или стоматологической процедуры. Нередко диагноз гемофилии ребенку устанавливают к 5–6 годам, обратив внимание на длительное посттравматическое кровотечение, длительное кровотечение во время стоматологического лечения или операции. К другим заметным симптомам легкого и умеренного течения гемофилии относятся непроходящие гематомы (синяки), частые носовые кровотечения, кровоточивость десен.
Для тяжелой формы гемофилии характерны эпизоды спонтанного кровотечения, которые приводят к кровоизлияниям различной локализации — в мягкие ткани, мышцы, суставы. При гемартрозах (кровоизлияние в полость сустава) возникает ограничение подвижности суставов, сопровождающееся острой болью и воспалением. У пациентов с тяжелой формой гемофилии спонтанные кровотечения чаще всего происходят в мышцы и суставы, однако могут затрагивать любой внутренний орган, включая почки, органы ЖКТ, головной мозг (гематурия, мелена, гематохезия, желудочно-кишечные кровотечения, внутричерепные кровотечения). Тяжелая форма гемофилии A обычно проявляется в раннем детском возрасте, диагноз чаще всего устанавливается к 2 годам ребенка. Обычными симптомами (при отсутствии лечения заболевания) здесь являются кровотечения из-за незначительных травм ротовой полости (прикусывание губ, языка, щек), подкожные гематомы, большие шишки после удара головой, спонтанные кровотечения (2–5 эпизодов в месяц).
Диагноз гемофилии A или B устанавливается на основании симптомов, истории личного и семейного анамнеза пациента, лабораторных исследований (общего анализа крови, коагулограммы, оценивающей состояние системы свертывания крови и активности ее белков, молекулярно-генетического исследования). Парам из группы риска по рождению ребенка с гемофилией необходимо получить генетическую консультацию на этапе планирования беременности. Определение конкретной мутации гена F8 (или F9) помогает не только выявить женщин-носительниц дефектного гена в конкретной семье, но и полезно для пренатальной диагностики гемофилии (амниоцентез, биопсия хориона).
Некоторым пациентам с легкой формой гемофилии назначается синтетический аналог вазопрессина — десмопрессин, повышающий VIII фактор свертывания крови (вводится внутривенно или интраназально), а также антифибринолитические средства, замедляющие распад факторов свертывания крови.
Примерно в 30 % случаев лечения тяжелых случаев гемофилии длительная заместитетельная терапия может приводить к изоиммунизации — образованию антител к вводимым факторам свертывания крови (иммунная система распознает вводимый фактор VIII как чужеродный). Этот процесс может сопровождаться аллергическими реакциями (разной степени тяжести), возрастает риск жизнеугрожающих кровотечений. В этом случае пациенту назначается альтернативное лечение — плазмаферез, иммунодепрессанты.
Гемохроматозы
Гемохроматоз — наследственное заболевание, характеризующееся нарушением обмена железа и его накоплением в тканях и органах.
Заболевание сопровождается повышенным всасыванием железа в желудочно-кишечном тракте и накоплением в печени, сердце, поджелудочной железе, суставах, гипофизе, что приводит к полиорганной недостаточности и развитию таких болезней, как цирроз печени, рак печени, диабет, болезни сердца и суставов.
Наследственный гемохроматоз вызывают мутации генов HFE, HFE2, HAMP, SLC40A1 и TfR2, он классифицируется в зависимости от возраста начала, генетической причины и способа наследования: гемохроматоз 1 типа, гемохроматоз 2 типа, гемохроматоз 3 типа, гемохроматоз 4 типа. Вторичный гемохроматоз ассоциирован с другими заболеваниями (не является наследственным) — анемией, хронической болезнью печени, инфекциями.
Гемохроматоз, редко возникающий в младенческом возрасте и не имеющий явной причины, называют неонатальным гемохроматозом. При этой форме заболевания избыточное накопление железа в тканях и органах начинается еще до рождения ребенка. Неонатальный гемохроматоз быстро прогрессирует и характеризуется поражениями печени, которые выявляют при рождении или в первые дни жизни. Дети с этим заболеванием часто рождаются недоношенными или имеют нарушения внутриутробного развития. Точная причина неонатального гемохроматоза неизвестна, есть версия, что он развивается в том случае, если иммунная система матери распознает клетки печени ребенка как чужеродные. Симптомы обычно включают гипогликемию, нарушения свертываемости крови, пожелтение кожных покровов и склер глаз, отеки. Диагноз устанавливается на основании признаков и симптомов, лабораторных и инструментальных исследований, биопсии печени. Лечение включает переливание крови, внутривенное введение иммуноглобулинов, трансплантацию печени.
Гемохроматоз 1 типа ассоциирован с мутациями в гене HFE, расположенном на коротком плече 6–й хромосомы, наследуется по аутосомно-рецессивному типу, является наиболее распространенным типом наследственного гемохроматоза и поражает в основном мужчин. Вероятность того, что ребенок унаследует мутацию в гене HFE от родителя составляет 25 %, вероятность того, что он станет носителем дефектного гена — 50 %.
Начальные симптомы заболевания обычно отмечаются в возрасте 40–60 лет и включают боль в животе, снижение полового влечения, усталость, слабость, боли в суставах, сухость кожи. В дальнейшем проявляются такие симптомы и осложнения, как изменение пигментации кожи (бронзовая кожа), выпадение волос на голове и туловище, аритмия, кардиомиопатия, хроническая сердечная недостаточность, сахарный диабет, гепатомегалия, цирроз печени, спленомегалия, атрофия яичек, аменорея (у женщин) и другие.
Диагноз устанавливается на основании симптомов заболевания, лабораторных исследований (уровень железа, ферритина, трансферрина, железосвязывающей способности сыворотки крови, маркеры функции печени, уровень глюкозы крови и др.), инструментальных исследований (рентгенография суставов, электрокардиография и эхокардиография сердца, УЗИ органов брюшной полости, МРТ печени и др.), биопсии печени, молекулярно-генетического исследования (в том числе для выявления родственников — имеющих гемохроматоз или носителей).
Лечение гемохроматоза направлено на удаление избытка железа из организма (флеботомия, хелатирование) с помощью забора крови (как при взятии анализов или донорстве крови, только в большем объеме) и специальных препаратов, образующих комплексное соединение с железом и способствующих его удалению из железосодержащих белков (дефероксамин).
Терапевтическая флеботомия сначала проводится 1–2 раза в неделю, поддерживающая — каждые 2–4 месяца. Вместе с этим проводится симптоматическое лечение сахарного диабета, патологий сердца, цирроза печени и других заболеваний, вызванных гемохроматозом. Пациентам запрещен прием препаратов железа и любых медикаментозных средств или комплексов, которые могут содержать железо и витамин C, запрещен алкоголь во избежание дальнейшего повреждения печени (если оно имеется), рекомендуется придерживаться диеты, исключающей продукты с высоким содержанием железа (красное мясо, яблоки, печень, шпинат).
Гемохроматоз 2 типа вызывают мутации в генах HFE2 или HAMP, он наследуется по аутосомно-рецессивному типу и проявляется чаще всего в детском возрасте. Для этого типа заболевания характерны следующие симптомы: высокие уровни ферритина и трансферрина, врожденный фиброз печени, артропатия, кардиомиопатия, генерализованная гиперпигментация кожи, мышечная слабость, гипогонадизм, задержка полового созревания, сахарный диабет, цирроз печени, остеопороз, спленомегалия, гепатомегалия и другие. Диагностика и лечение гемохроматоза 2 типа проводятся аналогично диагностике и лечению гемохроматоза 1 типа.
Гемохроматоз 3 типа вызывают мутации в гене TFR2, он наследуется по аутосомно-рецессивному типу. Симптомы обычно проявляются до 30 лет и, помимо вышеперечисленных, могут включать снижение уровня лимфоцитов и нейтрофилов в крови, красные или пурпурные пятна на коже. Диагностика и лечение гемохроматоза 3 типа проводятся аналогично диагностике и лечению гемохроматоза 1 и 2 типов.
Симптомы гемохроматоза 4 типа могут проявиться как в детском, так и во взрослом возрасте. Этот тип заболевания вызывают мутации гена SLC40A1, наследуется оно по аутосомно-доминатному типу. Вероятность того, что ребенок унаследует мутацию в гене SLC40A1 от родителя составляет 50 %, соответственно, вероятность того, что он станет носителем дефектного гена — тоже 50 %. Симптомы гемохроматоза 4 типа могут проявляться в виде повышенной утомляемости, слабости, болей в суставах, изменения пигментации кожи, затруднения дыхания, сердечной недостаточности, сахарного диабета, анемии, врожденного фиброза печени, цирроза печени, остеоартроза, катаракты. Диагностика и лечение гемохроматоза 4 типа проводятся аналогично диагностике и лечению гемохроматоза 1, 2 и 3 типов.
Наследственный гемохроматоз — заболевание с пониженной пенетрантностью (вероятностью того, что ген будет иметь любые фенотипические проявления) дефектного гена, т. е. у некоторых людей с мутациями гена HFE никогда не проявятся симптомы, при этом у их детей или других членов семьи с мутацией гена произойдет манифестация заболевания.
Мышечная дистрофия Дюшенна
Мышечная дистрофия Дюшенна — редкое наследственное заболевание, характеризующееся нарастающей мышечной слабостью с последующей атрофией мышц. Ассоциировано с мутацией гена DMD, расположенного на половой X-хромосоме (X-сцепленный рецессивный тип наследования).
Полипозные синдромы характеризуются наличием/развитием множественных полипов в различных отделах желудочно-кишечного тракта, но часто сопровождаются другими проявлениями. Некоторые полипозные синдромы неизбежно приводят к злокачественной трансформации полипов и развитию рака (например, САТК, АСАТК); другие - не связаны с развитием рака напрямую, но могут служить индикаторами повышенного риска возникновения некоторых кишечных или внекишечных опухолей.
Семейный аденоматоз толстой кишки (САТК, АСАТК)
• Фенотип: множественные аденоматозные полипы по всей толстой кишке, периампулярные дуоденальные полипы, полипы желудка, внекишечные проявления (десмоиды и т.д.).
• Тип наследования: аутосомно-доминантный, почти полная пенетрантность гена.
• Локализация гена: ген аденоматозного полипоза толстой кишки (АРС) локализован в хромосоме 5q21.
• Обычное течение заболевания: почти 100% развитие рака толстой кишки в возрасте 40-50 лет, в 3-12% случаев - периампулярный рак.
• Ассоциированные опухоли: рак толстой кишки, рак в резервуаре, периампулярная аденокарцинома, десмоидные опухоли, рак щитовидной железы.
• Варианты:
- Синдром Гарднера: остеомы, десмоидные опухоли, новообразования щитовидной железы, врожденная гипертрофия пигментного эпителия сетчатки.
- Синдром Турко: опухоли головного мозга.
- Аттенуированная форма САТК (АСАТК): более поздние проявления, более проксимальное расположение полипов.
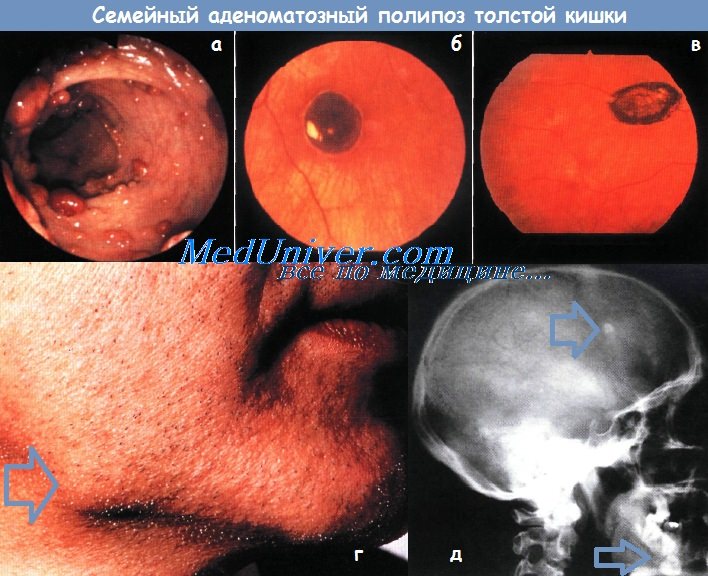
а - Колоноскопическая картина больного полипозом. Видно множество маленьких полипов.
б - Глазное дно при врожденной гипертрофии пигментного эпителия сетчатки.
У данного пациента это единственное поражение (по-липоз отсутствует).
в - Глазное дно больного с полипозом при врожденной гипертрофии пигментного эпителия сетчатки.
Одно из множественных поражений — светлый ореол с нечеткими очертаниями вокруг участка усиленной пигментации.
г - Остеома нижней челюсти у больного полипозом.
д - Рентгенограмма черепа больного полипозом. Видны остеома свода черепа и два таких же очага в нижней челюсти.
MYH-ассоциированный полипоз (МАП)
• Фенотип: часто не отличим от САТК, за исключением несколько меньшего числа полипов толстой кишки, внекишечные проявления присутствуют, но менее выражены, чем при САТК: полипы верхних отделов ЖКТ (=> периампулярный рак), остеомы, изменения зубов, наследственная гипертрофия пигментного эпителия сетчатки и др.
• Тип наследования: аутосомно-рецессивный, почти полная пенетрантность гена.
• Локализация гена: ген репарации MYH, хромосома 1р34-32.
• Обычное течение заболевания: диагноз МАП устанавливается в возрасте около 50 лет, почти в 100% случаев к 65 годам развивается рак.
• Ассоциированные опухоли: рак толстой кишки, периампулярная аденокарцинома, рак молочной железы, рак щитовидной железы.
• Консультация: оба родителя и все дети являются носителями гена.
Синдром Пейтца-Егерса
• Фенотип: гамартомные полипы ЖКТ, в частности его верхних отделов, отложение меланина в коже (например, около рта, на слизистой щек и т.д.).
• Тип наследования: аутосомно-доминантный с различной пенетрантностью гена.
• Локализация гена: LKB1/STK (хромосома 19р13) и другие гены.
• Обычное течение заболевания: у большинства больных бессимптомное, в редких случаях отмечаются синдромы обструкции и кровотечения.
• Ассоциированные опухоли: умеренно повышенный риск развития опухолей ЖКТ и опухолей внекишечной локализации.
Ювенильный полипоз
• Фенотип: гамартомные полипы, в 15% сочетающиеся с врожденными дефектами развития.
• Тип наследования: аутосомно-доминатный.
• Локализация гена: BMPR1A или SMAD-4 ген (хромосома 18q21) и другие гены.
• Обычное течение заболевания: средний возраст начала заболевания - 18 лет, наиболее частая локализация полипов - ректосигмоидный отдел; симптомы вариабельны: кровотечения из ЖКТ, инвагинация, выпадение прямой кишки, протеиндефицитная энтеропатия.
• Ассоциированные опухоли: значительно повышен риск развития колоректального рака.
• Диагностические критерии: > трех ювенильных полипов, полипоз всего ЖКТ, или любое количество полипов при наличии семейного анамнеза ювенильных полипов.
• Внимание: отдельные ювенильные полипы не малигнизируются.
Синдром Коудена
• Фенотип: синдром множественных гамартом из эктодермальных и в меньшей степени из эндодермальных элементов (трихолеммома — 80% случаев, макроцефалия - 40% случаев, полипоз ЖКТ - только 35% случаев, доброкачественные заболевания щитовидной и молочной желез).
• Тип наследования: аутосомно-доминантный, почти полная пенетрантность гена.
• Локализация гена: ген опухолевой супрессии PTEN, хромосома 10q23.
• Обычное течение заболевания: симптомы заболевания появляются к 20 годам.
• Ассоциированные опухоли: риск развития опухолей ЖКТ не повышен, в 10% случаев возникает рак щитовидной железы, в 30-50% - рак молочной железы.
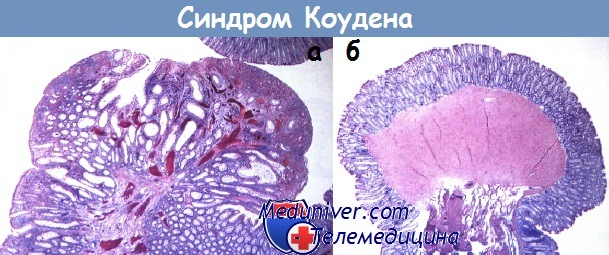
а - Синдром Коудена. Микроскопическая картина типичной гамартомы слизистой оболочки.
Пролиферация ветвистых фокально расширенных желез с умеренным количеством воспаленной стромы, которая может содержать пучки гладких мышц, фокальная эрозированная поверхность.
б - Второй тип гамартом при болезни Коудена — эквивалент лейомиомы мышечной пластинки слизистой оболочки.
Синдром Баннаяна-Райли-Рувалькаба (ранее синдром Рувалькаба-Майра-Смита)
• Фенотип: прогрессивный рост до/после рождения, макроцефалия, задержка умственного и психомоторного развития, другие аномалии; множественные гамартомные полипы ЖКТ; липомы; пигментные пятна на гениталиях.
• Тип наследования: аутосомно-доминантный, почти полная пенетрантность гена.
• Локализация гена: ген опухолевой супрессии PTEN, хромосома 10q23.
• Обычное течение заболевания: педиатрический аналог синдрома Коудена.
• Ассоциированные опухоли: риск развития колоректального рака, других опухолей ЖКТ и опухолей внекишечной локализации не повышен.
Синдром Кронкхайта-Канада
• Фенотип: диффузный полипоз всего ЖКТ (за исключением пищевода), эктодермальные аномалии (например, алопеция, ониходистрофия, гиперпигментация кожи).
• Тип наследования: аутосомно-доминантный.
• Локализация гена: ген опухолевой супрессии PTEN, хромосома 10q23.
• Обычное течение заболевания: диарея, белководефицитная энтеропатия, потеря веса, тошнота, рвота, анорексия, парестезии, тонико-клонические судороги, обусловленные электролитными нарушениями.
• Ассоциированные опухоли: повышенный риск развития рака желудка, толстой и прямой кишки (на момент установления диагноза отмечаются в 15% случаев).
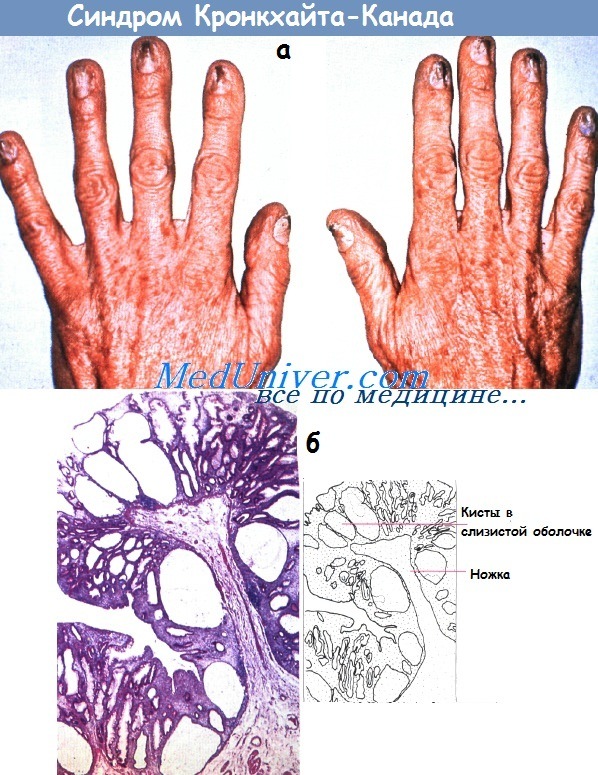
а - Ониходистрофия при синдроме Кронкхайта-Канада.
б - Гистологические проявления синдрома Кронкхайта-Канада.
Кистозные железы не диспластичны и не имеют стромальных элементов, позволяющих отличить их от других форм. Окраска гематоксилин-эозином (х 8).
Синдром гиперпластического полипоза
• Фенотип: множественные гиперпластические полипы всех отделов толстой и прямой кишки, включая крупные (> 1 см) полипы, локализованные прокси-мальнее сигмовидной кишки.
• Тип наследования: неизвестен.
• Локализация гена: неизвестна.
• Обычное течение заболевания: средний возраст 50-70 лет, специфические симптомы отсутствуют.
• Ассоциированные опухоли: повышенный риск развития колоректального рака (чаще проксимальных отделов).
Нодулярная лимфоидная гиперплазия
• Фенотип: множественные лимфоидные полипы всех отделов толстой и прямой кишки.
• Тип наследования: неизвестен.
• Локализация гена: неизвестна.
• Обычное течение заболевания: дети и взрослые, специфические симптомы отсутствуют, иногда сочетается с иммунодефицитом.
• Ассоциированные опухоли: риск развития колоректального рака не повышен.
Дополнительные исследования при полипозах кишечника
Генетическая консультация и тестирование => оценка индивидуального и семейного риска.
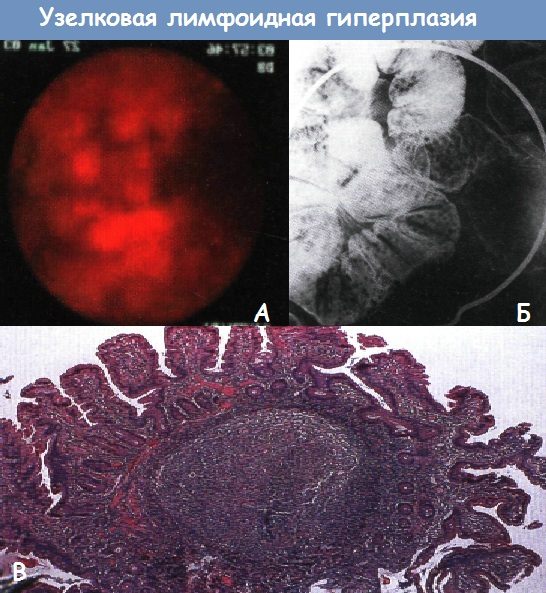
А - Капсульная эндоскопия. Лимфоидная гиперплазия у молодого пациента, неотягощенная другой патологией.
Б - Лимфоидная гиперплазия у пациента с вариабельным неклассифицируемым иммунодефицитом. Пероральная пневмоколонография. Множественные 1-2 мм рентгенопрозрачные дефекты наполнения в небольшом скоплении бария разделены нормальной гладкой слизистой оболочкой
В - Гистологический препарат подвздошной кишки с гиперплазией лимфатических узлов при иммунодефиците. Значительное увеличение пейеровых бляшек (терминальных центров) в подслизистом слое придает слизистой оболочке полиповидный вид
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Читайте также: