
Реферат на тему фенилкетонурия
Обновлено: 02.07.2024
Министерство здравоохранения и социального развития РФ
Государственное образовательное учреждение высшего профессионального образования
Санкт-Петербургская государственная химико-фармацевтическая академия
Выполнили студентки 340 группы
Фармацевтического факультета
Федорец ПолинаАндреевна
Исаченко Анастасия
Руководитель:
Спасенкова Ольга Михайловна
кандидат медицинских наук
доцент кафедры биохимии
Санкт-Петербург
2017
Содержание
Фенилкетонурия является редким, но тяжелым заболеванием, которое приводит к нарушению умственного развития человека в раннем возрасте. До 50-х гг. прошлого века она оставаласьмалоизученной и не поддавалась лечению. Впервые заболевание было описано в 1934 г Иваром Асбьёрном Фёллингом, и постепенно с развитием медицины и, в частности биохимии и генетики, у учёных сформировалось необходимое представление о причинах, механизме развития, а, следовательно, и о способах лечения фенилкетонурии. Тысячи детей были спасены от слабоумия, серьезных поражений печени и почек, а такжелетального исхода, который нередко влекло это заболевание.
Однако, на сегодняшний день некоторые вопросы остаются открытыми: например до какого возраста больной фенилкетонурией должен придерживаться диеты, возможны ли новые более эффективные методы ранней диагностики болезни, а также альтернативные методы лечения?
Все они могут и должны решаться с использованием комплексного подхода, на основе знаний иопыта медицины, генетики и биохимии. Биохимия занимает важное место и является неотъемлемой составляющей в системе медицинских наук. Изучение биохимических процессов человека, животных, растений оказывает содействие общему прогрессу научных знаний и развитию здравоохранения. Органические соединения является звеном, что скрепляет представления о живой и неживой природе в единую цепь. На основе знаний обэтих соединениях и процессах раскрываются тайны развития в материальном мире от простейших молекулярных форм к полимерным с присущими им функциями, вплоть до функций в живом организме. Все это определяет актуальность выбора темы работы.
Фенилкетонурия как результат нарушения метаболизма фенилаланина
ФЕНИЛКЕТОНУРИЯ - тяжелое наследственное заболевание, которое характеризуется главным образом поражением нервной системы.
Этиология и патогенез. В результате мутации гена, контролирующего синтез фенилаланингидроксилазы, развивается метаболический блок на этапе превращения фенилаланина в тирозин, вследствие чего основным путем преобразования фенилаланина становится дезаминирование и синтез токсических производных - фенилпировиноградной, фенил-молочной и фенилуксусной кислот. В крови и тканях значительно увеличивается содержание фвнилаланина (до 0,2 г/л и более при норме 0,01-0,02 г/л). Существенную роль в патогенезе болезни играет недостаточный синтез тирозина, который является предшественником катехоламинов и меланина. Заболевание наследуется по аутосомно-рецессивному типу.
Фенилкетонурия (ФКУ) - тяжелое наследственное заболевание, наступающее вследствие врожденного дефекта фермента, отвечающего в организме человека за нормальный обмен фенилаланина (одной из незаменимых аминокислот, входящих в состав белка).
При заболевании нарушаются обменные процессы, особенно важные для развивающегося мозга ребенка. В крови и других жидкостях организма накапливается в большом количестве фенилаланин и повышено образуются такие вещества как фенилпировиноградная, фенилмолочная и фенилуксусная кислоты, которые выделяются в повышенных количествах с мочой. Следствием нарушенного обмена в мозге является тяжелое психическое недоразвитие. Если не предпринято своевременное лечение, то больные на всю жизнь остаются глубокими инвалидами.
Поступающий в организм фенилаланин идет на построение белковой цепи или превращается в тирозин. Отсутствие в печени фермента фенилаланингидроксидазы препятствует нормальному превращению фенилаланина пищи в тирозин. Поэтому фенилаланин используется лишь при синтезе белка, а избыток накапливается в клетках печени и попадает в кровоток, где количество фенилаланина является токсичным для клеток мозга. Почки не справляются с его реабсорбцией, в результате чего он выводится с мочой. Именно наличие этого фенилкетона в моче дало основание назвать соответствующее патологическое состояние фенилкетонурией.

Варианты ФКУ
Классическая фенилкетонурия (ФКУ) описана А.Folling.,1934г.
Заболевание наследуется аутосомно-рецессивно и вызвано мутацией гена, локализующегося в длинном плече 12 хромосомы.
В основе болезни лежит дефицит фермента фенилаланин-4-гидроксилазы, обеспечивающего превращение фенилаланина в тирозин. В результате метаболического блока происходит значительное накопление в тканях и жидкостях больного организма фенилаланина и таких его производных, как фенилпировиноградная, фенилмолочная, фенилуксусная кислоты, фенилэтиламин, фенилацетилглютамин, и др.
- Нарушение в обмене белков, липо- и гликопротеидов;
- Расстройства транспорта аминокислот;
- Нарушение метаболизма гормонов;
- Нарушение обмена моноаминовых нейромедиаторов (катехоламинов и серотонина);
- Нарушение функции печени - диспротеинемия, генерализованная гипераминоацидемия, повышение ДФА, метаболический ацидоз, нарушение окислительной и белковосинтезирующей функции клеточных органелл.
Частота классической ФКУ среди новорожденных по данным массового скрининга в среднем колеблется от 1:5000 до 1:10000 по разным регионам России.
Впервые атипичная ФКУ описана I.Smith, 1974г. Заболевание связано с дефицитом дигидроптеридинредуктазы.
Заболевание наследуется аутосомно-рецессивно. Генный дефект локализуется в коротком плече 4 хромосомы, участке 4р 15.3.
В результате недостаточности дигидроптеридинредуктазы нарушается восстановление активной формы тетрагидробиоптерина, участвующего в качестве кофактора в гидроксилировании фенилаланина, тирозина, и триптофана.
Частота заболевания составляет 1:100000 новорожденных.
Рано начатое лечение способствует нормализации фенилаланина в крови, однако не предупреждает появление клинической симптоматики, которая развивается в начале второго полугодия жизни. Фенилкетонурию 2 называют диеторезистентной ФКУ.
Этот вариант болезни описал S. Kaufman в 1978 г. Заболевание наследуется аутосомно-рецессивно и связано с недостаточностью 6-пирувоилтетрагидроптерин синтетазы, участвующей в процессе синтеза тетрагидробиоптерина. Развивающиеся при этом расстройства сходны с нарушениями, наблюдаемыми при ФКУ 2.
Частота болезни составляет 1:30000 новорожденных. Фенилкетонурия 3 также диеторезистентна.
Другие варианты ФКУ:
Эти формы ФКУ связаны с нарушением альтернативных путей обмена фенилаланина. Формируется метилминдальная ацидурия и парагидроскифенилуксусная ацидурия.
Заболевание развивается у потомков женщин, страдающих ФКУ и не получающих диету в зрелом возрасте. Патогенез мало изучен, предполагается, что он сходен с патогенезом остальных форм ФКУ. Тяжесть поражения плода коррелирует с уровнем фенилаланина в плазме матери. Так как эмбрион особенно чувствителен к тератогенным воздействиям , рекомендуется начинать диету еще до наступления беременности. В суточном рационе использовать менее 15-20 мг/кг фенилаланина. При это важно избегать дефицита незаменимых аминокислот.
При рождении больные фенилкетонурией не отличаются от других новорожденных. Манифестация ФКУ происходит обычно в возрасте 2-6 месяцев.
Клинические проявления
По мере прогрессирования болезни могут наблюдаться эпилептиформные приступы - развернутые судорожные и бессудорожные типа кивков, поклонов, вздрагиваний, кратковременных отключений сознания. Гипертония отдельных групп мышц проявляется своеобразной "позой портного" (поджатые ноги и согнутые руки). Могут наблюдаться гиперкинезы, атаксия, тремор рук, иногда парезы по центральному типу. Дети нередко белокурые со светлой кожей и голубыми глазами, у них часто отмечаются экзема, дерматиты. Обнаруживается склонность к артериальной гипотензии.
Первыми проявлениями болезни служат:
- вялость ребенка, отсутствие интереса к окружающему;
- повышенная раздражительность, беспокойство;
- судорожные эквиваленты: спонтанный рефлекс Моро, спонтанный рефлекс Бабинского, сосательные автоматизмы, приапизм у мальчиков, атетозные движения;
- заплесневелый, мышиный, волчий запах мочи и пота.
Дигностика фенилкетонурии
Чрезвычайно важно установить диагноз в доклинической стадии или по крайней мере не позднее 2-го месяца жизни, когда могут проявиться первые признаки болезни. Для этого всех новорожденных обследуют по специальным профаммам скрининга, выявляющего повышение концентрации фенилаланина в крови уже в первые недели жизни. Оптимальные сроки обследования новорожденных - 5-14-день жизни . Каждого ребенка, у которого обнаруживаются признаки задержки развития или минимальная неврологическая симптоматика, необходимо обследовать на патологию обмена фенилаланина. Используют микробиологический и флюорометрический методы определения концентрации фенилаланина в крови, а также пробу Фелинга на фенилпировиноградную кислоту в моче (прибавление нескольких капель 5% раствора треххлористого железа и уксусной кислоты к моче больного приводит к появлению зеленой окраски пятна на пеленке). Эти и другие подобные методы относятся к категории ориентировочных, поэтому при положительных результатах требуется специальное обследование с использована ем точных количественных методов определения содержания фенилаланина в крови ц моче (хроматография аминокислот, использование аминоанализаторов и др.), которое осуществляется централизованными биохимическими лабораториями.
Дети требуют специального наблюдения и лечения в медико-генетических центрах (кабинетах поликлиник).
Дифференциальный диагноз проводят с внутричерепной родовой травмой, внутриутробными инфекциями.
ФКУ может быть диагностирована на основе обнаружения следующих признаков:
o стойкой гиперфенилаланинемии (более 240 ммоль/л);
o вторичного дефицита тирозина;
o экскреции фенилкетонов с мочой (проба Феллинга на экскрецию фенилпировиноградной кислоты).
o микробиологический тест Гатри;
o метод флюоресцирующих антител (лабораторный комплекс "Флюороскан", позволяющий проводить 800 проб в час);
o метод тонкослойной хроматографии.
Оптимальные сроки обследования новорожденных - доношенных, зрелых - 5-6 день жизни; недоношенных, незрелых, больных - 10-14 день жизни.
Уровень фенилаланина не превышает 200 ммоль/л (1-3 мг%) - норма;
Уровень фенилаланина составляет 200-500 ммоль/л (3-10 мг%) - гиперфенилаланинемия. В эту группу входят дети с транзиторной гиперфенилаланинемией, вследствие незрелости ферментных систем печени и больные ФКУ. За данной группой проводится наблюдение по следующему плану: если в течение 6 недель при еженедельном исследовании уровень фенилаланина остается менее 500 ммоль/л, контроль за уровнем фенилаланина крови проводят до 1 года первоначально каждые 3 месяца, а затем каждые 6 мес. жизни. При уровне более 500 ммоль/л назначается диетотерапия.
Уровень фенилаланина превышает 500 ммоль/л (более 10 мг%), диагностируется ФКУ и с момента постановки диагноза назначается диетотерапия.
В настоящее время разрабатываются и внедряются молекулярно-генетические методы диагностики генного дефекта при ФКУ. Прямая диагностика мутантного гена проводится с помощью синтетических олигонуклеотидных зондов, этот метод пригоден для дородовой диагностики ФКУ и выявления гетерозиготного носительства.
Помимо молекулярно-генетического анализа, выявление гетерозигот может осуществляться биохимическими тестами после нагрузки фенилаланином в дозе 25 мг/кг.
Диеторезистентные формы ФКУ диагностируют при помощи:
o исследования биоптеринов мочи ;
o перорального нагрузочного теста с тетрагидробоиптерином (через 4-6 часов после однократной дачи нагрузки в дозе 7,5 мг/кг массы тела происходит резкое снижение и нормализация уровня фенилаланина в крови с одновременным повышением уровня тирозина);
o исследования активности дигидроптеридинредуктазы и 6-пирувоилтетрагидроптерин синтетазы в культуре кожных фибробластов, эритроцитах, гепатоцитах.
Наследственность
Лечение фенилкутонурии и прогноз
Если ничего не предпринимать, фенилкетонурия приводит к весьма тяжелым последствиям - развивается олигофрения. К счастью, этот трагический исход можно предотвратить, если поставить правильный диагноз при рождении. В наши дни это легко осуществимо в ходе массового обследования (скрининга) новорожденных с целью выявления ФКУ.
Главным способом лечения является диетотерапия, ограничивающая поступление в организм фенилаланина; приступить к ней нужно немедленно после установления диагноза. При ранней диагностике это гарантирует нормальное нервно-психическое развитие ребенка.
Диетотерапия, как единственный эффективный метод лечения ФКУ, должна применятся с первых месяцев жизни ребенка, тогда поражение мозга не разовьется. Важно ограничить количество потребляемого фенилаланина таким образом, чтобы обеспечить его поступление в организм в количествах, необходимых и достаточных для роста и развития, но предотвратив его накопление в жидкостях тела. Кроме диетотерапии необходим постоянный медицинский контроль за умственным и физическим развитием ребенка.
Очень важно! Применение диетотерапии на позднем этапе не вернет ребенку нормального интеллекта. Дети, у которых это заболевание не диагностируют сразу при рождении, а выявляют по умственной отсталости, не могут быть излечены.
По достижении 12-14 лет такие дети могут переходить на нормальное питание и никаких признаков отравления фенилаланином у них не наблюдается. Однако женщина, которая в детстве переболела ФКУ, должна снова перейти на диету и употреблять только продукты с пониженным содержанием фенилаланина перед зачатием, и оставаться на этой диете во время беременности и кормления грудью. Если она не сделает этого, то ее ребенок подвергается риску замедленного физического и умственного развития, даже если его отец не является носителем гена ФКУ.
Диетотерапия
Единственным лечением, способным предотвратить развитие слабоумия или уменьшить его степень, является диета, исключающая поступление в организм фенилаланина сверх того минимального количества, которое необходимо для образования собственных белков организма и его роста. Поэтому смысл диетического лечения сводится к резкому ограничению естественного белка с пищей.
Для такого ограничения приходится полностью исключить из питания ребенка такие богатые белками продукты как мясо, колбасы, рыбу, бульоны, яйца, творог, сыр, мучные изделия, каши из естественных круп, фасоль, орехи, шоколад. Меню для детей составляется из фруктов, овощей, крахмальных изделий, жиров, со строгим учетом содержания в них фенилаланина.
Назначают белковые гидролизаты (цимогран, лефанолак, берлофен, гипофенат) или аминокислотные смеси, лишенные фенилаланина, которые становятся главными продуктами питания, обеспечивающими потребность в белке: "Лофенолак", "Фенилфри" (США), "Берлофен", "Апонти", "Гипофенат" у детей до 4-5 лет и "Нофелан" - у детей старше 5 лет. Белковые гидролизаты вводят с фруктовыми и овощными соками, пюре, супами.
Лечение проводят под контролем содержания фенилаланина в крови, добиваясь поддержания его уровня в пределах 0,03-0,04 г/л.
Строгое ограничение белков животного происхождения требуется на протяжении первых 2-3 лет жизни. Наиболее рационально отменять диетическое лечение в возрасте 7-8 лет.
1.Наком (комбинация карбиДОФА и левоДОФА) - доза 100-375 мг/сутки в течение 3-4 недель, перерыв между курсами 1,5-2 месяца;
2.Лево-дофа - доза 10-15 мг/кг в сутки;
3.5-окситриптофан - доза 10 мг/кг сутки.
При диеторезистентных формах лечение включает назначение тетрагидробиоптерина - доза 10-20 мг/кг в сутки.
Профилактика фенилкетонурии
1. Выявление гетерозиготных носителей. Большое значение имеет специальное наблюдение за семьями риска, т. е. за такими семьями, где уже имелись дети с фенилкетонурией. Новорожденные из этих семей должны быть подвергнуты обязательному биохимическому исследованию и при показаниях к раннему лечению.
2. Внедрение программ массового скрининга новорожденных для раннего выявления ФКУ и своевременного назначения диетотерапии. Выявление и лечение детей по программам массового скрининга также позволяет предупредить развитие тяжелой психической инвалидности.
3. Пренатальная диагностика: Предложен ДНК-зонд для пренатальной диагностики фенилкетонурии в семьях высокого риска.
Ребенок с фенилкетонурией выглядит при рождении здоровым. Отставание психического развития может происходить постепенно и стать очевидным лишь через несколько месяцев . Установлено, что нелеченный ребенок теряет около 50 баллов IQ к концу первого года жизни. Отставание психического развития обычно довольно выражено, и большинство детей нуждаются в социальной помощи.
Содержание
•Актуальность
•Этиология, патогенез
•Клиническая картина
•Возможности диагностики
•Лечение
•Заключение
•Список литературы
Фенилкетонурия (ФКУ)
Работа содержит 1 файл
ФЕНИЛКЕТОНУРИЯ.doc
ГОУ ВПО
Орловский государственный университет
Медицинский институт
Кафедра педиатрии, детской хирургии и медицинской генетики
Зав. кафедрой: Васина Г. И.
Выполнила: студентка 5 группы
4 курса Завьялова Е. С.
Проверила: асс. Ставцева С.Н.
Орел, 2009
Содержание реферата
- Актуальность
- Этиология, патогенез
- Клиническая картина
- Возможности диагностики
- Лечение
- Заключение
- Список литературы
Фенилкетонурия (ФКУ)
- Фенилкетонурия (ФКУ) является наиболее распространенной аминоацидопатией. Частота ФКУ среди новорожденных по данным массового скрининга в различных странах составляет в среднем 1:10000, однако значительно варьирует в зависимости от популяции: от 1:4560 в Ирландии, до 1:100.000 в Японии. (см. таблицу)
Ребенок с фенилкетонурией выглядит при рождении здоровым. Отставание психического развития может происходить постепенно и стать очевидным лишь через несколько месяцев . Установлено, что нелеченный ребенок теряет около 50 баллов IQ к концу первого года жизни. Отставание психического развития обычно довольно выражено, и большинство детей нуждаются в социальной помощи.
- В связи с тяжестью клинических проявлений и возможностью профилактического лечения ФКУ рекомендована для выяв ления среди новорожденных. Ранняя диагностика позволяет своевременно начать лечение и предотвратить инвалидизацию больного.
- Лечение и реабилитация больных ФКУ требует значительных финансово-экономических затрат. Это обусловлено высокой себестоимостью продуктов лечебного питания, а также усилиями по социальной реабилитации больного.
- Помимо этого, об актуальности заболевания, свидетельствует проблема материнской ФКУ, лишь недавно появившаяся в России. Наблюдается высокая частота умственной отсталости среди потомства женщин, страдающих ФКУ и не получающих диету в зрелом возрасте.
- Скудная клиническая картина в раннем возрасте, постепенное развитие патологических изменений, приводит к серьезным затруднениям в ранней диагностике этого заболевания. В тоже время, диетотерапия больных с ФКУ должна находиться под тщательным динамическим контролем. А психоневрологическая реабилитация больных (медикаментозная, физиолечение, медико-педагогическая) требует индивидуального подхода. Педиатры, в чьи обязанности входит реабилитация больных ФКУ, встречаются с ней в повседневной практике нечасто. Поэтому необходимо ознакомить врачей с особенностями течения и лечением ФКУ.
Табл. Заболеваемость ФКУ по данным массового скрининга
Схема патогенеза фенилкетонурии
По Mc Kusick выделяется несколько типов фенилкетонурии.
Фенилкетонурия I
Классическая фенилкетонурия. Была описана А. Folling в 1934 г. Заболевание наследуется аутосомно- рецессивно и вызвано мутацией гена ФАГ, локализующегося в длинном плече 12 хромосомы. В гене ФКУ выявлено 12 различных гаплотипов . При этом 90% всех генов ФКУ ассоциировано с четырьмя гаплотипами, из них гаплотип 3 характеризует около 38% всех генов ФАГ, а гаплотип 2 - около 20% .
В основе болезни лежит дефицит фермента фенилаланин - 4 - гидроксилазы (ФАГ), обеспечивающей превращение фенилаланина в тирозин. В результате метаболического блока происходит значительное накопление в тканях и жидкостях больного организма фенилаланина и таких его производных, к ак фенилпировиноградная, фенилмолочная, фенилуксусная кислоты, фенилэтиламин, фенилацетилглютамин и др.
По мнению различных авторов в патогенезе ФКУ имеют значение следующие обстоятельства: прямое токсическое действие на центральную нервную систему фенилаланина и его производных, нару шения в обмене белков, липо- и гликопротеидов, расстройства транспорта аминокислот, нарушен ие метаболизма гормонов и др., а также перинатальные факторы. В последнее время все большее знач ение в патогенезе ФКУ придается нарушениям обмена моноаминовых нейромедиаторов (катехаламинов и серотонина). Известна исключительно важная роль этих медиаторов в функционировании центральной нервной системы. Исследования показали, что у больных резко снижено содержан ие конечных продуктов их метаболизма (гомованилиновой кислоты и 5- оксииндолуксусной кислоты) в крови, моче и цереброспинальной жидкости.
Определенное значение в генезе церебральных расстройств могут играть нарушения функции печени. У большинства больных ФКУ при обследовании обнаруживаются различные биохимические и морфологические изменения, свидетельствующие о заинтерсованности этого органа в патологическом процессе: диспротеинемия, генерализованная гипераминацидемия, повышение показателя дифениламиновой реакции, компенсированный метаболический ац идоз, признаки белковой и жировой дистрофии печени с нарушением окислительной и белоксинтезирую щей функции клеточных органелл.
Фенилкетонурия II
Впервые атипичная ФКУ была описана I. Smith в 1974 г. S. Kaufman et al. В 1975 г. обнаружили дефицит дигидроптеридинредуктазы при этом состоянии.
Заболевание наследуется аутосомно- рецессивно. Генный дефект локализуется в коротком плече 4 хромосомы, участке 4р15.3 . В результате недостаточности дигидроптеридинредуктазы нарушается восстановление активной формы тетрагидробиоптерина, участвующего в качестве кофактора в гидроксилировании фенилаланина, тирозина и триптофана. Вследствие этого развиваются метаболические блоки на путях превращения фенилаланина в тирозин, а также образования предшественников нейромедиаторов катехоламинового и серотонинового ряда L-дофы и 5- окситриптофана , что подтверждается резким снижением содержания в тканях и жидкостях больного организма (в том числе в мозге и цереброспинальной жидкости) их конечных продуктов - гомованилиновой и 5- оксиндолуксусной кислот.
Существенным для патогенеза заболевания является снижение уровня фолатов в сыворотке крови, эритроцитах и цереброспинальной жидкости. Это объясняется тесной взаимосвязью обмена фолатов и биоптерина, в частности участием дигидроптеринредуктазы в метаболизме тетрагидрофолиевой кислоты.
Фенилкетонурия III
Этот вариант болезни впервые описан S. Kaufman et al. в 1978 г. Заболевание наследуется аутосомно- рецессивно и связано с недостаточностью 6-пирувоилтетрагидроптерин синтазы, участвующей в процессе синтеза тетрагидробиоптерина из дигидронеоптерин трифосфата . Ключевую роль в патогенезе играет дефицит тетрагидробиоптерина. Развивающиеся при этом расстройства сходны с нарушениями, наблюдаемыми при ФКУ II.
Другие варианты ФКУ
В последние годы стали известны другие формы атипичной ФКУ, связанные с дефицитом тетрагидробиоптерина.
Недостаточность гуанозин 5- трифосфат циклогидролазы (S. Kaufman et al., 1987) описана по крайней мере у пяти больных. Этот фермент катализирует первую ступень синтеза тетрагидробиоптерина и при его дефиците в моче обнаруживается крайне низкая концентрация всех птеринов.
N. Blau и соавторы (1989) сообщили о новом варианте атипичной ФКУ - примаптеринурии - у двух детей с легкой гиперфенилаланинемией. Энзиматический дефект пока не известен. В моче обнаруживается в больших количествах продукт изомеризации биоптерина-7 - изобиоптерин (примаптерин) и некоторые другие его производные. Соотношение неоптерин/биоптерин у больных значительно повышено. Нагрузка тетрагидробиоптерином нормализует концентрацию фенилаланина в сыворотке и уровень неоптерина в моче, резко повышает экскрецию биоптерина и примаптерина. Отличием от других атипичных форм ФКУ является нормальная концентрация в цереброспинальной жидкости нейромедиаторных метаболитов - гомованилиновой и 5-оксииндолуксусной кислот.
Материнская ФКУ
В процессе изучения ФКУ было обращено внимание на высокую частоту умственной отсталости среди потомства женщин, страдающих ФКУ и не получающих диету в зрелом возрасте. Это состояние получило наименование материнской ФКУ . Патогенез патологии мало изучен , однако предполагается, что он сходен с патогенезом остальных форм ФКУ. Тяжесть поражения плода коррелирует с уровнем фенилаланина в плазме матери. Причем в связи с накоплением этой аминокислоты в плаценте, ее содержание в организме плода оказывается выше, чем у матери. Тем не менее прямое токсическое действие фенилаланина точно не подтверждено. Появление признаков патологии у потомства не зависит от наличия или отсутствия умственной отсталости у женщин и не связано с развитием у детей ФКУ. Есть данные, что тяжесть заболевания у больных ФКУ, родившихся от матерей, также страдающих этой болезнью, не отличается от степени поражения их сибсов, не унаследовавших ФКУ .
Фенилкетонурия I
Манифестация ФКУ происходит на первом году жизни, обычно в возрасте 2-6 мес. Первыми проявлениями болезни служат вялость ребенка, отсутствие интереса к окружающему, иногда повышенная раздражительность, беспокойство, срыгивания, нарушение мышечного тонуса (чаще мышечная гипотония), судороги, признаки аллергического дерматита. Появляется характерный “мышиный” запах. Отчетливо формируется задержка статикомоторного и психоречевого развития. При отсутствии лечения умственная отсталость достигает, как правило, глубокой степени.
Ранним симптомом заболевания может стать рвота, иногда настолько сильная, что ее ошибочно расценивают как проявление пилоростеноза. В более старшем возрасте нелеченные дети становятся гиперактивными, осуществляют бесцельные движения, ритмические покачивания, у них определяется атетоз.
При физикальном исследовании обращает на себя внимание то, что ребенок выглядит более белокурым, чем его здоровые сиблинги: у него светлая кожа и голубые глаза. У некоторых больных появляется себорейная или экзематоидная кожная сыпь, обычно умеренно выраженная и исчезающая по мере роста ребенка. От них исходит необычный запах фенилуксусной кислоты, который характеризуют как заплесневелый, мышиный или волчий. Не встречается характерных неврологических изменений , однако у большинства детей определяются гипертонус и повышение глубоких сухожильных рефлексов. Около 1/4 детей страдают судорогами и более чем у 50% появляются изменения на ЭЭГ. Часто у нелеченных детей определяют микроцефалию, выступающую верхнюю челюсть с широко расставленными зубами, гипоплазией эмали, отставание роста. Клинические проявления классической фенилкетонурии редко встречаются в странах, в которых эффективно действует программа неонатального скрининга на это заболевание.
Фенилкетонурия II
В клинической картине ФКУ II преобладает тяжелая умственная отсталость, судороги, признаки повышенной возбудимости, сухожильная гиперрефлексия, мышечная дистония, спастический тетрапарез. Течение болезни прогрессирующее и нередко приводит к смерти в 2- З- летнем возрасте. Больные, как правило, выявляются в результате массового скрининга новорожденных на ФКУ. Уровень фенилаланина в крови у этих детей увеличен, иногда может превышать 1200 мкмоль/л. Появление клинической симптоматики, как правило развивается в начале второго полугодия жизни, несмотря на диетотерапию.
Фенилкетонурия III
Клиническая картина болезни напоминает ФКУ II и включает тяжелую умственную отсталость, микроцефалию, спастический тетрапарез. Для подтверждения диагноза требуется :
- Для учеников 1-11 классов и дошкольников
- Бесплатные сертификаты учителям и участникам
Фенилкетонурия — наследственное заболевание, связанное с нарушением аминокислотного обмена, обусловленное недостаточностью печеночных ферментов, участвующих в метаболизме фенилаланина до тирозина, с последующим тяжелым поражением ЦНС проявляющимся, в частности, в виде нарушения умственного развития. Это одно из немногих наследственных заболеваний, поддающихся успешному лечению.
Фенилкетонурия является заболеванием с аутосомно-рецессивным характером наследования. Это означает, что для развития клинических признаков фенилкетонурии ребенок должен унаследовать по одной дефектной копии гена от обоих родителей, являющихся гетерозиготными носителями мутантного гена.
Фенилкетонурия может быть спровоцирована следующими факторами:
Близкородственные браки, при которых, помимо иных патологий, повышается вероятность рождения ребенка с этим заболеванием;
Мутация гена (т.е. его изменение), произошедшая по тем или иным причинам в области локализации 12 хромосомы.
Новорожденные с фенилкетонурией не имеют клинических признаков заболевания. Дети, страдающие фенилкетонурией, рождаются с правильно сформированным и нормально функционирующим головным мозгом. Такое положение связано с тем, что головной мозг формируется у плода, и все биохимические процессы плода зависят от обмена веществ матери. Однако при фенилкетонурии биохимические нарушения головного мозга грудничка начинаются сразу же после рождения. Обычно манифестация фенилкетонурии у детей происходит в возрасте 2-6 месяцев. С началом кормления в организм ребенка начинает поступать белок грудного молока либо его заменителей, что приводит к развитию первых, неспецифических симптомов – вялости, иногда – беспокойства и гипервозбудимости, срыгивания, мышечной дистонии, судорожного синдрома. Одним из ранних патогномоничных признаков фенилкетонурии служит упорная рвота, которая нередко ошибочно расценивается как проявление пилоростеноза.
У детей с фенилкетонурией, не получающих лечения, выявляется микроцефалия (уменьшение размеров черепа и головного мозга), прогнатия, позднее (после 1,5 лет) прорезывание зубов, гипоплазия эмали. Отмечается задержка речевого развития, а к 3-4 годам выявляется глубокая олигофрения и практически полное отсутствие речи.
Клинические проявления фенилкетонурии II типа характеризуются тяжелой степенью умственной отсталости, повышенной возбудимостью, судорогами, спастическим тетрапарезом, сухожильной гиперрефлексией. Прогрессирование заболевание может приводить к гибели ребенка в возрасте 2-З лет.
При фенилкетонури III типа развивается триада признаков: микроцефалия, олигофрения, спастический тетрапарез.
Таким образом при данном заболевании имеется множество психопатологических нарушений.
У 92-96 % людей проявляются тяжелые степени умственной отсталости. У 3-4 % имеется легкая недостаточность интеллекта. И только у 0,2-0,3 % не выявляется никаких отклонений. Ситуация ухудшается в первые несколько лет жизни ребенка, после чего процесс стабилизируется. Дети не проявляют никаких эмоций – их мимика маловыразительна. Такие дети не общаются со сверстниками и родителями, они импульсивны, имеют определенные стереотипы движения, строят гримасы, повторяют слова (эхолалия).
Главным способом лечения ФКУ является диетотерапия, ограничивающая поступление в организм пищевого белка и фенилаланина до минимальной возрастной потребности. В пищевой рацион больных входят овощи, фрукты, соки, а также специальные малобелковые продукты (саго, хлеб, вермишель, крупка, приготовленная на крахмальной основе). Однако в период интенсивного роста и развития ребенка поступление белка в организм должно быть достаточным, так как дефицит его незамедлительно отразится на процессе формирования всех органов и систем. Поэтому из рациона новорожденного нельзя полностью исключить материнское молоко. Для коррекции питания детям даются белковые гидролизаты, лишенные фенилаланина, но содержащие все другие необходимые аминокислоты. Больные нуждаются в дополнительном введении витаминов, особенно группы В, минеральных веществ и микроэлементов.
Методика обучения и воспитания детей с фенилкетонурией
Данная категория больных нуждается не только в обязательной своевременной медицинской, но и коррекционно-педагогической помощи. В основе занятий с ними должен лежать комплекс коррекционно-развивающих и психологических приемов, позволяющих преодолеть недоразвитие речи и нарушения познавательной деятельности, недостатки эмоционально-волевой сферы, поведения и негативных черт личности. Необходимо развивать у детей высшие психические функции, формировать познавательный интерес, целенаправленность и настойчивость в деятельности. Без этого невозможны реализация их интеллектуальных возможностей и адаптация в социальной среде.
Огромную роль в реабилитации больных ФКУ играет логопедическое воздействие. С момента поступления больных в отделение логопед в течение двух недель проводит совместно с врачом-психиатром диагностику речевых и интеллектуальных нарушений у детей, составляет перспективный план коррекционной работы, подбирает методы и приемы обучения строго в индивидуальном порядке. Затем в соответствии с планом и расписанием в течение всего срока госпитализации с детьми проводятся ежедневные индивидуальные и 2-3 раза в неделю подгрупповые занятия. С детьми старшего дошкольного возраста параллельно с педагогом логопед ведет подготовку к обучению в школе. Практически у всех больных детей отмечается задержка речевого развития или общее недоразвитие речи всех уровней (I-III уровни речевого развития), иногда осложненные дизартрией, алалией, заиканием. При постоянной коррекционной работе речевые диагнозы детей изменяются в лучшую сторону. Логопеды должны уделять большое внимание фонетической и лексико-грамматической сторонам речи детей, развитию связной речи; сенсорному восприятию, формированию пространственно-временных представлений, представлений об окружающей действительности и отражением их в речи.
1. Установление контакта.
Например, с помощью педагога ребенок выполняет несложный рисунок (яблоко, цветок, солнце) и всегда получает похвалу. Кроме того, в конце каждого занятия проводится короткая и интересная для ребенка игра (по его выбору), которая назначается как бы за хорошее поведение на занятии. Через некоторое время (обычно через 1-2 мес.) у ребенка возникает устойчивое положительное отношение к занятиям, появляется желание посмотреть на педагога, чтобы увидеть его реакцию на свои действия, устанавливается рабочий контакт с ним.
2. Введение оценки деятельности.
В беседе ребенка знакомят со школьными оценками - "отлично" ("5") и "плохо" ("2"). "Пятерку" всегда пишут красным цветом, а "двойку" - черным, что придает им особую аффективную значимость. Сначала оцениваются только легкие задания, с которыми ребенок хорошо справляется, например рисование простых форм или предметов. Вскоре детей уже не удовлетворяет устная похвала за проделанную работу, и педагог выставляет им "школьную" отметку.Важнейшей задачей педагога будет подготовка ребенка к преодолению ситуации неуспеха.
3. Развитие самоконтроля.
Детям предлагают проверить работы других учеников (списывание слов из букваря печатными буквами), в которых либо были допущены ошибки, либо ошибки отсутствовали. Детям говорят, что они должны выставить оценку за работу - "пять" или "два" или оставить работу без оценки. Это задание выполняется с большой охотой - дети проверяют по букварю написание слов (Маша, рама, ушла) и ставят оценки. Затем предлагается оценивать так свою работу, не случается, чтобы ребенок поставил себе плохую оценку. Но спустя некоторое время ребенок уже начинает адекватно оценивать работу
4. Развитие самооценки.
Как следствие проделываемой в течение первого полугодия работы у детей начинает формироваться адекватная самооценка в сфере учебных действий и заданий (о самооценке личностных качеств ниже). Это проявляется, прежде всего, в том, как они выполняют задание на исследование уровня притязаний.
5. Преодоление эгоцентризма.
Работа в направлении преодоления эгоцентризма начинается с чтения и обсуждения рассказов, которые требуют оценки ситуации и сопереживания персонажу. Примерами таких рассказов являются "Горькое лекарство" и "Самая красивая".Итогом такой работы становится то, что к концу года дети становятся внимательнее и добрее к родным, менее холодны. Они уже различают состояние и настроение матери (усталая, грустная, веселая) и учитывают это в своих поступках. Отношения с родителями становятся более теплыми.
6. Изменение родительских установок на воспитание ребенка.
Присутствуя на занятиях, выполняя с ребенком домашние задания, родители начинают видеть его как бы с другой стороны. Если раньше они считали своей основной задачей лечение ребенка, то теперь начинают понимать, что одного лечения недостаточно. Ребенка нужно учить, так как он отстает от сверстников. Его нужно воспитывать, поскольку, если даже делать скидку на болезнь, его личность имеет целый ряд негативных черт.Совместные занятия, игры ведут и к лучшему взаимопониманию родителей и ребенка. Таким образом, меняется место ребенка в семье - на него начинают смотреть не как на больного, а как на будущего ученика
Фенилкетонурия – это наследственное нарушение аминокислотного обмена, обусловленное недостаточностью печеночных ферментов, участвующих в метаболизме фенилаланина до тирозина. Ранними признаками фенилкетонурии служат рвота, вялость или гиперактивность, запах плесени от мочи и кожи, задержка психомоторного развития; типичные поздние признаки включают олигофрению, отставание в физическом развитии, судороги, экзематозные изменения кожи и др. Скрининг новорожденных на фенилкетонурию проводится еще в родильном доме; последующая диагностика включает молекулярно-генетическое тестирование, определение концентрации фенилаланина в крови, биохимический анализ мочи, ЭЭГ, МРТ головного мозга. Лечение фенилкетонурии заключается в соблюдении специальной диеты.
МКБ-10
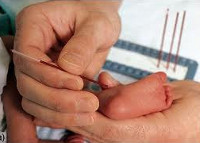
Общие сведения
Фенилкетонурия (болезнь Феллинга, фенилпировиноградная олигофрения) – врожденная, генетически обусловленная патология, характеризующаяся нарушением гидроксилирования фенилаланина, накоплением аминокислоты и ее метаболитов в физиологических жидкостях и тканях с последующим тяжелым поражением ЦНС. Фенилкетонурия впервые описана А. Феллингом в 1934 г.; встречается с частотой 1 случай на 10 000 новорожденных.
В неонатальном периоде фенилкетонурия не имеет клинических проявлений, однако поступление фенилаланина с пищей вызывает манифестацию заболевания уже в первом полугодии жизни, а в дальнейшем приводит к тяжелым нарушениям развития ребенка. Именно поэтому пресимптоматическое выявление фенилкетонурии у новорожденных является важнейшей задачей неонатологии, педиатрии и генетики.
Причины фенилкетонурии
Фенилкетонурия является заболеванием с аутосомно-рецессивным характером наследования. Это означает, что для развития клинических признаков фенилкетонурии ребенок должен унаследовать по одной дефектной копии гена от обоих родителей, являющихся гетерозиготными носителями мутантного гена.
Чаще всего к развитию фенилкетонурии приводит мутация гена, кодирующего фермент фенилаланин-4-гидроксилазу и расположенного на длинном плече 12 хромосомы (локус12q22-q24.1). Это, так называемая, классическая фенилкетонурия I типа, составляющая 98% всех случаев заболевания. Гиперфенилаланинемия может достигать 30 мг% и выше. При отсутствии лечения данный вариант фенилкетонурии сопровождается глубокой умственной отсталостью.
Кроме классической формы, различают атипичные варианты фенилкетонурии, протекающие с той же клинической симптоматикой, но не поддающиеся коррекции диетотерапией. К ним относятся фенилкетонурия II типа (недостаточность дегидроптеринредуктазы), фенилкетонурия III типа (дефицит тетрагидробиоптерина) и другие, более редкие варианты. Вероятность рождения ребенка, больного фенилкетонурией, повышается при заключении близкородственных браков.
Патогенез
В основе классической формы фенилкетонурии лежит недостаточность фермента фенилаланин-4-гидроксилазы, участвующего в конверсии фенилаланина в тирозин в митохондриях гепатоцитов. В свою очередь, производный тирозина – тирамин является исходным продуктом для синтеза катехоламинов (адреналина и норадреналина), а дийодтирозин – для образования тироксина. Кроме этого, результатом метаболизма фенилаланина служит образование пигмента меланина.
Наследственная недостаточность фермента фенилалаиин-4-гидроксилазы при фенилкетонурии приводит к нарушению окисления фенилаланина, поступающего с пищей, в результате чего его концентрация в крови (фенилаланинемия) и спинномозговой жидкости значительно возрастает, а уровень тирозина соответственно падает. Избыточное содержание фенилаланина устраняется путем повышенной экскреции с мочой его метаболитов - фенилпировиноградной, фенилмолочной и фенилуксусной кислот.
Нарушение обмена аминокислот сопровождается нарушением миелинизации нервных волокон, снижением образования нейромедиаторов (дофамина, серотонина и др.), запускающими патогенетические механизмы задержки умственного развития и прогредиентное слабоумие.
Симптомы фенилкетонурии
Новорожденные с фенилкетонурией не имеют клинических признаков заболевания. Обычно манифестация фенилкетонурии у детей происходит в возрасте 2-6 месяцев. С началом кормления в организм ребенка начинает поступать белок грудного молока либо его заменителей, что приводит к развитию первых, неспецифических симптомов – вялости, иногда – беспокойства и гипервозбудимости, срыгивания, мышечной дистонии, судорожного синдрома. Одним из ранних патогномоничных признаков фенилкетонурии служит упорная рвота, которая нередко ошибочно расценивается как проявление пилоростеноза.
У детей с фенилкетонурией, не получающих лечения, выявляется микроцефалия, прогнатия, позднее (после 1,5 лет) прорезывание зубов, гипоплазия эмали. Отмечается задержка речевого развития, а к 3-4 годам выявляется глубокая олигофрения (идиотия) и практически полное отсутствие речи.
Клинические проявления фенилкетонурии II типа характеризуются тяжелой степенью умственной отсталости, повышенной возбудимостью, судорогами, спастическим тетрапарезом, сухожильной гиперрефлексией. Прогрессирование заболевание может приводить к гибели ребенка в возрасте 2-З лет. При фенилкетонури III типа развивается триада признаков: микроцефалия, олигофрения, спастический тетрапарез.
Диагностика
В настоящее время диагностика фенилкетонурии (а также галактоземии, врожденного гипотиреоза, адрено-генитального синдрома и муковисцидоза) входит в программу неонатального скрининга, осуществляемого всем новорожденным. Основные и дополнительные методы диагностики:
- Скрининг-тест. Проводится на 3-5 день жизни доношенного и 7 день жизни недоношенного ребенка путем забора образца капиллярной крови на специальный бумажный бланк. При обнаружении гиперфенилаланемии более 2,2 мг% ребенка направляют к детскому генетику для повторного обследования.
- Биохимические исследования. Для подтверждения диагноза фенилкетонурии проверяется концентрация фенилаланина и тирозина в крови, определяют активность печеночных ферментов (фенилаланингидроксилазы), выполняется биохимическое исследование мочи (определение кетоновых кислот), метаболитов катехоламинов в моче и др.
- Неврологическое обследование. Дополнительно проводится ЭЭГ и МРТ головного мозга, осмотр ребенка детским неврологом.
- Пренатальная диагностика. Генетический дефект при фенилкетонурии может быть обнаружен еще на этапе беременности в ходе инвазивной пренатальной диагностики плода (хорионбиопсии, амниоцентеза, кордоцентеза). В остальных случаях окончательный диагноз выставляется по результатам ДНК-диагностики после рождения.
Дифференциальный диагноз фенилкетонурии проводят с внутричерепной родовой травмой новорожденных, внутриутробными инфекциями, другими нарушениями обмена аминокислот.
Лечение фенилкетонурии
Основополагающим фактором в лечении фенилкетонурии является соблюдение диеты, ограничивающей поступление белка в организм. Лечение рекомендуется начинать при концентрации фенилаланина >6 мг%. Для грудных детей разработаны специальные смеси - Афенилак, Лофенилак; для детей старше 1 года – Тетрафен, Фенил-фри; старше 8 лет - Максамум-ХР и др. Основу диеты составляют низкобелковые продукты - фрукты, овощи, соки, белковые гидролизаты и аминокислотные смеси. Расширение диеты возможно после 18 лет в связи с возрастанием толерантности к фенилаланину. В соответствии с российским законодательством обеспечение лиц, страдающих фенилкетонурией, лечебным питанием, должна осуществляться бесплатно.
Больным назначается прием минеральных соединений, витаминов группы В и др.; по показаниям - ноотропные средства, антиконвульсанты. В комплексной терапии фенилкетонурии широко используется общий массаж, ЛФК, иглорефлексотерапия. Атипичные формы фенилкетонурии, не поддающиеся лечению диетой, требуют назначения гепатопротекторов, противосудорожных средств, заместительной терапии леводопой, 5-гидрокситриптофаном.
Дети, страдающие фенилкетонурией, находятся под наблюдением участкового педиатра и психоневролога; нередко нуждаются в помощи логопеда и дефектолога. Необходим тщательный мониторинг нервно-психического статуса детей, контроль уровня фенилаланина в крови и показателей электроэнцефалограммы.
Прогноз и профилактика
Проведения массового скрининга на фенилкетонурию в неонатальном периоде позволяет организовать раннюю диетотерапию и предотвратить тяжелые церебральные повреждения, нарушения функции печени. При раннем назначении элиминационной диеты при классической фенилкетонурии прогноз развития детей хороший. При поздно начатом лечении прогноз в отношении умственного развития неблагоприятный.
Профилактика осложнений фенилкетонурии заключается в проведении массового скрининга новорожденных, раннего назначения и длительного соблюдения диетического питания.
С целью оценки риска рождения ребенка с фенилкетонурией предварительное генетическое консультирование должны пройти супружеские пары, уже имеющие больного ребенка, состоящие в кровнородственном браке, имеющие родственников с данным заболеванием. Женщины с фенилкетонурией, планирующие беременность, должны соблюдать строгую диету до зачатия и во время беременности для исключения повышения уровня фенилаланина и его метаболитов и нарушения развития генетически здорового плода. Риск рождения ребенка с фенилкетонурией у родителей-носителей дефектного гена, составляет 1:4.
Читайте также: