
Неонатальный скрининг новорожденных реферат
Обновлено: 05.07.2024
Невозможно недооценить значимость скрининга новорожденных. Ведь большинство генетических патологий абсолютно не проявляются при рождении и на протяжении первых месяцев жизни: малыш может выглядеть здоровым, но при этом иметь наследственный дефект. И только со временем проявляются симптомы, позволяющие диагностировать болезнь. Раннее распознавание заболеваний и незамедлительное лечение в доклинический период болезни дает возможность предотвратить стремительное развитие недуга и избежать серьезных осложнений.
Содержание работы
I.Введение…………………………………………………………..1
II.Определение…………………………………………………….2
III. Заболевания, на которые проводится скрининг……………2
IV.Технология проведения неонатального скрининга…………3
V.Список литературы…………………………………………….5
Файлы: 1 файл
Неонатальный скрининг.docx
Тема: Неонатальный скрининг новорожденных
III. Заболевания, на которые проводится скрининг……………2
IV.Технология проведения неонатального скрининга…………3
Скрининг – это массовое обследование новорожденных детей, проводимое с целью выявления генетических заболеваний. Специальные лабораторные тесты позволяют определить некоторые наследственные болезни до появления их симптомов.
Невозможно недооценить значимость скрининга новорожденных. Ведь большинство генетических патологий абсолютно не проявляются при рождении и на протяжении первых месяцев жизни: малыш может выглядеть здоровым, но при этом иметь наследственный дефект. И только со временем проявляются симптомы, позволяющие диагностировать болезнь. Раннее распознавание заболеваний и незамедлительное лечение в доклинический период болезни дает возможность предотвратить стремительное развитие недуга и избежать серьезных осложнений.
Неонатальный скрининг проводят в родильном доме, однако некоторым детям по разным причинам его переносят на более поздние сроки, и тогда в амбулаторных условиях необходимо довести до родителей всю важность данного обследования и провести его.
III. Заболевания, на которые проводится скрининг
* Врожденный гипотиреоз – внутриутробное поражение зачатка щитовидной железы. Заболевание ведет к нарушению вырабатывания тиреоидных гормонов и, как следствие, к задержке развития роста и формирования нервной системы ребенка. Девочки страдают от гипотиреоза вдвое чаще, чем мальчики. Если болезнь своевременно диагностирована, то прием гормональных препаратов может полностью остановить ее развитие;
*Фенилкетонурия – генетическое заболевание, характеризующееся дефицитом фермента, который способен расщепить аминокислоту фенилаланин. Накопление в крови этой аминокислоты и ее метаболитов в моче приводит к сильнейшей интоксикации, повреждению головного мозга, серьезным неврологическим нарушениям и умственной отсталости. Практически вся белковая пища содержит аминокислоту фенилаланин, поэтому основным лечением является соответствующая диета. Специфического питания необходимо придерживаться длительное время – порядка первых десяти лет жизни. При своевременно проведенном неонатальном скрининге новорожденного и вовремя начатом лечении дети, страдающие фенилкетонурией, не отстают от своих сверстников в умственном развитии;
*Адреногенитальный синдром – наследственная дисфункция коры надпочечников. Заболевание обусловлено нарушением вырабатывания гормона, влияющего на обменный процесс. В крови новорожденного уже на первой неделе жизни начинают накапливаться вещества, оказывающие пагубное влияние на сердечно-сосудистую систему, работу почек и формирование половых органов. Гормональное лечение проводится на протяжении всей жизни;
*Муковисцидоз – самое частое генетическое заболевание, характеризующееся мутацией гена, в результате чего происходит поражение желез внешней секреции. Болезнь приводит к тяжелым нарушениям функций органов дыхания и желудочно-кишечного тракта. Неонатальный скрининг новорожденных позволяет выявить недуг и назначить своевременное медикаментозное лечение;
*Гелактоземия – генетически обусловленное нарушение обмена веществ, характеризующееся дефицитом фермента, который превращает галактозу в глюкозу. В связи с этим происходит тяжелое нарушение нервной системы, печени и других органов. Основное лечение – специфическая диета, исключающая молочные продукты.
IV.Технология проведения неонатального скрининга
Проведение неонатального скрининга на наследственную патологию основано на определении дефектов ферментов, участвующих в обмене белков и углеводов, в сухом пятне крови на специальной фильтровальной бумаге (тест-бланке). Эти исследования проводят в медико-генетической консультации (центре), куда направляются образцы крови новорождённых одновременно на фенилкетонурию, врождённый гипотиреоз, адреногенитальный синдром, галактоземию, муковисцидоз.
Обязательным условием точности диагностики является тщательная пропитка кровью пятна на тест-бланке.
Забор образцов крови новорождённым в роддоме, в отделении выхаживания недоношенных или патологии новорождённых в детских больницах осуществляется специально подготовленной медсестрой. Кровь берут из пятки новорождённого через 3 ч после кормления: у доношенного ребёнка - на 4-й день жизни, у недоношенного - на 7-й день.
Определение термина "неонатальный скрининг", причины массового обследования детей в период новорождённости. Диетотерапия как эффективный метод лечения классической фенилкетонурии. Технология проведения неонатального скрининга на наследственную патологию.
| Рубрика | Медицина |
| Вид | реферат |
| Язык | русский |
| Дата добавления | 11.04.2018 |
| Размер файла | 17,0 K |
Студенты, аспиранты, молодые ученые, использующие базу знаний в своей учебе и работе, будут вам очень благодарны.
Министерство здравоохранения Российской Федерации
Федеральное государственное бюджетное образовательное учреждение высшего образования
Неонатальный скрининг новорожденных
Неонатальный скрининг проводят в родильном доме, однако некоторым детям по разным причинам его переносят на более поздние сроки, и тогда в амбулаторных условиях необходимо довести до родителей всю важность данного обследования и провести его.
Фенилкетонурия (ФКУ) - наследственное заболевание, в основе которого лежит нарушение обмена аминокислоты фенилаланина, которая, накапливаясь в крови и спинномозговой жидкости, вызывает поражение нервной системы. Частота ФКУ среди новорождённых 1 на 5000-10 000 (в России - 1 на 6950). Отставание в развитии ребёнка выявляется во втором полугодии жизни. Примерно у 60% больных отмечают идиотию, у остальных - менее выраженные умственные нарушения. Раннее выявление заболеваний у новорождённых, своевременное и правильное лечение таких больных с первых дней жизни предупреждает задержку умственного развития детей.
Врождённый гипотиреоз - одно из наиболее часто встречающихся заболеваний щитовидной железы вследствие нарушения синтеза её гормонов. Его частота составляет 1 случай на 4000-5000 новорождённых. В России ежегодно рождается 400 детей с врожденным гипотиреозом. В основе заболевания лежит полная или частичная недостаточность тиреоидных гормонов, приводящая к задержке развития всех органов и систем. В первую очередь страдает ЦНС и интеллект. При поздней диагностике и несвоевременном лечении дети становятся инвалидами с полной утратой способности к обучению, трудоспособности и к социальной адаптации. Своевременно начатое лечение тиреоидными гормонами предотвращает развитие умственной отсталости. Эффективность лечения зависит от срока постановки диагноза, так как уже в первые месяцы жизни наступают необратимые изменения в умственном развитии и росте скелета.
Неонатальный скрининг позволяет диагностировать гипотиреоз в первый месяц жизни ребёнка.
Адреногенитальный синдром - наследственное заболевание, обусловленное снижением активности фермента, участвующего в выработке гормонов надпочечника (кортизола и альдостерона). Распространённость, по данным разных авторов, колеблется от 1:5000 до 1:20 000 новорождённых. Клинические проявления зависят от того, на каком уровне блокируются ферменты. Наиболее тяжёлой, опасной для жизни является сольтеряющая форма, частота которой 1:27 000. Болезнь начинается в первую неделю жизни ребёнка, протекает остро, с выраженным обезвоживанием, падением артериального давления, судорогами и требует немедленного проведения реанимационных мероприятий с целью коррекции водно-электролитного баланса. При отсутствии адекватной терапии больные новорождённые умирают на 1-2-м месяце жизни. Для лечения назначают заместительную гормональную терапию (глюко- и минералкортикоиды).
Галактоземия - наследственное заболевание, связанное с невозможностью использования организмом углевода молока - галактозы. Частота болезни - 1 случай на 15 000-20 000 новорождённых. В основе её лежит отсутствие или резкое снижение активности ферментов, которые в процессе обмена веществ превращают галактозу молока в глюкозу. Вследствие неполного расщепления промежуточные продукты обмена оказывают токсическое воздействие на организм. Болезнь проявляется в виде тяжёлого поражения печени, нервной системы, глаз и других органов. Значительная часть больных, не получающих адекватной терапии, умирает в грудном возрасте; у других уже в первом полугодии жизни формируется тяжёлая инвалидизирующая патология: катаракта, цирроз печени, задержка нервнопсихического развития. При ранней постановке диагноза и своевременно начатом лечении сохраняется нормальный интеллект, не появляются нарушения глаз и печени. В настоящее время разработано и успешно применяется патогенетическое лечение диетой. Так как молоко (материнское и коровье) содержит лактозу, которая под действием ферментов расщепляется до галактозы и глюкозы, то с первых дней жизни, с момента установления диагноза, ребёнок должен быть переведён на безмолочное питание. Следует немедленно прекратить грудное вскармливание! неонатальный скрининг наследственный патология
Технология проведения неонатального скрининга. Проведение неонатального скрининга на наследственную патологию основано на определении дефектов ферментов, участвующих в обмене белков и углеводов, в сухом пятне крови на специальной фильтровальной бумаге (тест-бланке). Эти исследования проводят в медико-генетической консультации (центре), куда направляются образцы крови новорождённых одновременно на фенилкетонурию, врождённый гипотиреоз, адреногенитальный синдром, галактоземию, муковисцидоз.
Обязательным условием точности диагностики является тщательная пропитка кровью пятна на тест-бланке.
Забор образцов крови новорождённым в роддоме, в отделении выхаживания недоношенных или патологии новорождённых в детских больницах осуществляется специально подготовленной медсестрой. Кровь берут из пятки новорождённого через 3 ч после кормления: у доношенного ребёнка - на 4-й день жизни, у недоношенного - на 7-й день.
Алгоритм действий медсестры при взятии образцов крови:
- вымыть руки (гигиенический уровень), надеть перчатки;
- вымыть пятку ребёнка;
- протереть пятку стерильной салфеткой, смоченной 70% спиртом, промокнуть её сухой стерильной салфеткой;
- проколоть пятку стерильным одноразовым скарификатором;
- снять первую каплю крови стерильным сухим тампоном;
- мягко надавить на пятку для получения второй капли крови;
- приложить перпендикулярно тест-бланк к капле крови и пропитать его кровью насквозь;
- аналогичным образом нанести на тест-бланк 6-8 капель, вид пятен крови должен быть одинаковым с обеих сторон.
- высушить тест-бланк в горизонтальном положении на чистой обезжиренной поверхности не менее 2 ч без применения тепловой обработки и попадания прямых солнечных лучей;
- упаковать тест-бланки в чистый конверт таким образом, чтобы пятна крови не соприкасались.
Оформление сопроводительной документации. После забора образцов крови медсестра разборчиво записывает шариковой ручкой на тест-бланке, не затрагивая пятен крови, следующие сведения:
- наименование учреждения, в котором произведён забор образцов крови;
-фамилия, имя, отчество матери ребёнка;
-адрес выбытия матери ребёнка;
-порядковый номер тест-бланка с образцом крови;
-дата и номер истории родов;
-дата взятия образца крови;
-состояние ребёнка (здоров/болен);
- доношенный/недоношенный (срок гестации);
- масса тела ребёнка;
- фамилия, имя, отчество лица, осуществлявшего забор крови.
Список используемых источников
1. Неонатология : Учебн. пособие : В 2 т. / Н.П.Шабалов. -- Т. I. -- 3-е изд., испр. и доп. -- М. : МЕДпресс-информ, 2004. -- 608 с. : илл.
2. Сестринский уход за новорожденным в амбулаторно-поликлинических условиях.Под ред. Д.И. Зелинской. 2010г.
Подобные документы
Генетический скрининг как выявление в популяции лиц с определенным генотипом, который обуславливает заболевание. Система мероприятий по неонатальному скринингу. Основные требования к программам неонатального скрининга на наследственным болезням.
презентация [96,0 K], добавлен 08.10.2014
Характеристика лечебно-профилактического учреждения в целом и структурного подразделения отделения новорожденных. Основные качественные и количественные показатели деятельности отделения новорожденных. Проведение неонатального аудиологического скрининга.
аттестационная работа [2,7 M], добавлен 08.05.2016
Массовое обследование новорожденных детей на наследственные заболевания, которое проводится в родильных домах. Раннее выявление фенилкетонури, врожденного гипотериоза, адреногенитального синдрома, галактоземии, муковисцитоза и их своевременное лечение.
презентация [1,9 M], добавлен 10.11.2014
Понятие, основные причины и факторы развития рака молочной железы как наиболее распространенного злокачественного заболевания и ведущей причины смертности среди женщин. Сущность и методы проведения скрининга, его значение в диагностике этого заболевания.
презентация [306,5 K], добавлен 19.06.2014
Технология высокопроизводительного скрининга биообъектов как ключевой этап поиска и разработки лекарственных средств. Понятие, сущность, функции, ступени организации биообъектов. Особенности планирования биологических испытаний синтезированных соединений.
реферат [27,5 K], добавлен 01.06.2010
Оценка концентрации маркерных белков альфа-фетопротеина и бета-субъединицы ХГЧ в сыворотке женщин во время беременности. Схема пренатального скрининга. Метод для проведения скринингового теста – иммунофлуоресценция. Динамика выявления болезни Дауна.
презентация [3,2 M], добавлен 28.03.2016
Факторы, которые приводят к женскому и мужскому бесплодию. Обследование мужчины, состоящего в бесплодном браке. Объективное обследование женщины, которая не может забеременеть. Инфекционный и гормональный скрининг. Инструментальные методы обследования.

Разработка программы скрининга началась в шестидесятых годах прошлого века, когда Роберт Гатри создал технологию тестирования сухих отпечатков крови на фильтровальной бумаге. Первым заболеванием, которое стало кандидатом для массовой диагностики, была фенилкетонурия, так как ее раннее выявление и коррекция питания способны предотвратить развитие тяжелых неврологических нарушений. Затем к скринингу добавилось еще несколько заболеваний: врожденный гипотиреоз, ВДКН, галактоземия и муковисцидоз. Тандемная масс-спектрометрия (ТМС) позволила значительно расширить список заболеваний, добавив к болезням обмена веществ гемоглобинопатии, спинальную мышечную атрофию, тяжелый комбинированный иммунодефицит и др.
Таблица 1 | Рекомендуемая The American College of Obstetricians and Gynecologists (ACOG) скрининг-панель для врожденных заболеваний
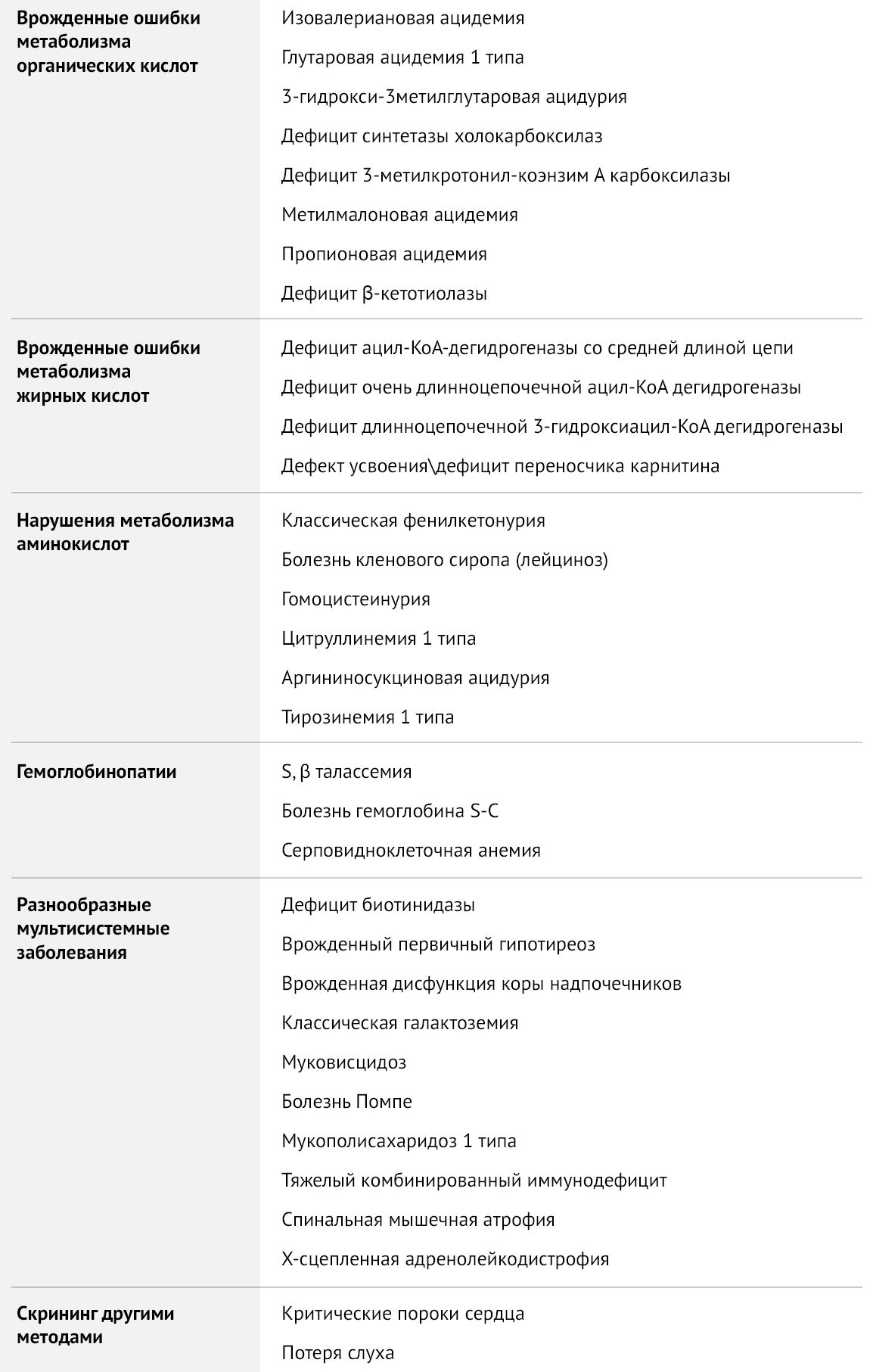
В данной статье будут рассмотрены два скрининга, доступные в нашей стране: обязательный, включающий тестирование на пять заболеваний (врожденный гипотиреоз, ВДКН, фенилкетонурия, галактоземия, муковисцидоз) и расширенный скрининг на наследственные нарушения метаболизма.
Фенилкетонурия (в современной классификации ― ФАГ-зависимая ФКУ) обусловлена мутацией гена фенилаланингидроксилазы и относится к числу аминокислотных аминоацидопатий. В норме фенилаланин (ФА) путем реакций гидроксилирования превращается в тирозин, однако в случае мутации вышеназванного гена активность превращающего фермента снижается, создается дефицит тирозина одновременно с избытком ФА, образующего токсичные метаболиты (фенилацетат, фенилпируват, фениллактат). Снижение образования тирозина влечет за собой нарушение синтеза гормонов щитовидной железы, нейротрансмиттеров и пигментов меланоцитов, а избыток ФА приводит к дисбалансу аминокислот в тканях мозга, обусловленному торможением их всасывания в желудочно-кишечном тракте или нарушением реабсорбции из почечных канальцев, нарушению образования или стабилизации полирибосом, снижению синтеза миелина, норадреналина и серотонина. Также за счет конкурентного ингибирования создается дефицит тирозиназы, что в совокупности с дефицитом тирозина приводит к снижению образования меланина и гипопигментации.
Основной проблемой пациентов с ФКУ являются нарушения функции ЦНС: от сонливости, вялости, отсутствия аппетита в период манифестации в 2–6 месяцев до тяжелых нарушений психомоторного развития в будущем; нередко развиваются атаксия, гиперкинезы, тремор рук, парезы по центральному типу. Единственный способ предотвратить развитие вышеназванных нарушений — назначение гипофенилаланиновой диеты с момента рождения с поддержанием низкого уровня фенилаланина в течение всей жизни.

Рисунок 1 | Интерпретация результатов исследования на наличие фенилкетонурии
ВДКН обусловлена дефицитом ферментов и транспортных белков, участвующих в биосинтезе кортизола. Наиболее часто встречается дефицит 21-гидроксилазы, что в свою очередь приводит к дефициту кортизола и альдостерона и ответному увеличению секреции АКТГ и гиперплазии коры надпочечников. В условиях дефицита фермента происходит значительное накопление предшественников гормонов, что приводит к увеличению синтеза тестостерона, не зависящего от 21-гидроксилазы. В итоге у пациента формируется надпочечниковая недостаточность и гиперандрогения. Гормональным маркером дефицита 21-гидроксилазы является уровень 17-гидроксипрогестерона (17-ОНП), определяемый в рамках неонатального скрининга. Результат трактуется как положительный, если при двукратном тестировании образца уровень 17-ОНП у доношенных новорожденных составляет ≥ 20 нг/мл. У недоношенных детей при заборе крови на 7–8 сутки после рождения скрининговый результат трактуется как положительный при следующих уровнях 17-ОНП: на сроке 23–32 недели гестации ― ≥ 65 нг/мл; на сроке 33–36 недель гестации ― ≥ 40 нг/мл.
Врожденный гипотиреоз в большинстве случаев вызван дефектами самой щитовидной железы (первичный гипотиреоз). Причины первичного врожденного гипотиреоза можно в широком смысле классифицировать как неспособность щитовидной железы нормально развиваться (дисгенезия) или неспособность структурно нормальной щитовидной железы производить нормальные количества гормона (дисгормоногенез). Дисгенезия щитовидной железы, охватывающая весь спектр агенеза, гипоплазии и эктопии, является наиболее частой причиной врожденного гипотиреоза. В то время как это заболевание остается наиболее частой причиной врожденного гипотиреоза, частота возникновения дисгормоногенеза за последние несколько десятилетий увеличилась. В то время как на дисгормоногенез приходится только 15 % врожденного гипотиреоза, диагностированного в первые дни скрининга новорожденных, у 30–40 % младенцев, прошедших скрининг по современным протоколам, имеется эктопическая щитовидная железа, соответствующая одной из форм дисгормоногенеза. В отличие от дисгенезии щитовидной железы, при которой моногенная причина присутствует только у небольшого количества пациентов, дисгормоногенез часто возникает из-за генетического дефекта на каком-либо этапе синтеза тиреоидных гормонов.
Учитывая разнообразие функций тиреоидных гормонов в организме человека, врожденный гипотиреоз характеризуется разнообразием клинических проявлений с поражением всех органов и систем. При отсутствии своевременного лечения на первый план выходит задержка психомоторного и речевого развития, затем наступают отставание в физическом развитии и задержка полового развития. Основной задачей скрининга является наиболее раннее выявление детей с подозрением на врожденный гипотиреоз.

Рисунок 2 | Интерпретация результатов исследования на наличие врожденного гипотиреоза
Галактоземия — аутосомно-рецессивное наследственное нарушение обмена углеводов, при котором в организме накапливается избыток галактозы и ее метаболитов. В норме галактоза образуется в результате гидролиза лактозы в кишечнике либо в процессе ферментных реакций, обмена гликопротеинов и гликолипидов. Галактоза является материалом для образования клеточных мембран, нервной ткани, нервных окончаний и т. д. В результате ферментных реакций она превращается в глюкозу, и именно дефицит галактозо-1-фосфатуридилтрансферазы лежит в основе патогенеза данного заболевания. Метаболиты галактозы обладают повреждающим действием. Так, галактитол проникает в хрусталик глаза, приводя к повышению осмотического давления, электролитным нарушениям и денатурации белка с формированием катаракты. Другие метаболиты обладают гепато-, нейро- и нефротоксическим действиями, а также вызывают гемолиз эритроцитов. Тормозящее влияние метаболитов галактозы на углеводный обмен приводит к гипогликемии.

Рисунок 3 | Интерпретация результатов исследования на наличие галактоземии
Муковисцидоз — аутосомно-рецессивное заболевание, связанное с мутацией гена МВТР (трансмембранного регулятора муковисцидоза). МВТР является хлорным каналом, мутации гена которого нарушают не только транспорт, но и секрецию ионов хлора. При затруднении их прохождения через клеточную мембрану увеличивается реабсорбция натрия железистыми клетками, нарушается электрический потенциал просвета, что вызывает изменение электролитного состава и дегидратацию секрета желез внешней секреции. В результате выделяемый секрет становится чрезмерно густым и вязким. Поражаются все экзокринные железы организма: печень, поджелудочная железа, мочеполовая система, но наиболее ярко муковисцидоз проявляет себя со стороны органов дыхания, провоцируя бронхообструкцию, дыхательную и сердечную недостаточность, легочную гипертензию.

Рисунок 4 | Интерпретация результатов исследования на наличие муковисцидоза
Органические ацидемии — группа аутосомно-рецессивных наследственных заболеваний обмена, в основе патогенеза которых лежит дефицит ферментов, участвующих в метаболизме белков, что приводит к повышению уровня кетоновых тел, обладающих токсическим действием на различные органы и ткани, в частности, на ЦНС. Данные заболевания манифестируют уже в стадии декомпенсации, как правило, в период с первой недели до первого года жизни. Триггерами служат стресс, длительное голодание, инфекционные заболевания, иммунизация, реже — чрезмерное употребление белковой пищи. Проявляются преимущественно неврологической симптоматикой: нарушение сознания вплоть до комы, эпилептические приступы, нарушение мышечного тонуса, у детей старшего возраста — нарушения психоречевого развития, атаксия, очаговые неврологические симптомы, синдром Рейе (острая печеночная недостаточность, сочетающаяся с энцефалопатией), психические расстройства.
Нарушения окисления жирных кислот — врожденный дефект метаболизма из-за нарушения либо митохондриального β-окисления, либо транспорта жирных кислот с использованием карнитинового транспортного пути. Проявления зависят от нарушения метаболизма конкретной кислоты, но все они имеют общие черты и требуют схожей тактики лечения. В периоде новорожденности метаболические нарушения проявляются тяжелой кардиомиопатией, гипокетотической гипогликемией, дисфункцией печени в первые несколько дней или недель жизни, часто заканчиваясь летально. В младенческом и детском возрасте характерны эпизоды летаргии и рвоты, развивается дисфункция печени и гипокетотическая гипогликемия, энцефалопатия, что может привести к внезапной младенческой смерти. У подростков и во взрослом возрасте дебютируют эпизодическим рабдомиолизом, мышечной слабостью, миалгией. Лечение включает отказ от голодания, симптоматическую терапию развившихся осложнений и включение в рацион добавок, если это необходимо.
Аминоацидопатии
Болезнь кленового сиропа (она же лейциноз) — наследственное заболевание, обусловленное дефицитом дегидрогеназы кетокислот с разветвленной цепью и нарушением метаболизма лейцина, изолейцина, валина (аминокислоты с разветвленной цепью, АКЦР). Повышение уровня АКЦР и их метаболитов, в частности, кетокислот, приводит к кетоацидозу, атрофии ткани головного мозга, нарушению окислительного фосфорилирования в дыхательной цепи митохондрий. Избыток лейцина обладает нейротоксическим эффектом, вызывая дисфункцию астроцитов, апоптоз нейронов и блокируя транспорт через гематоэнцефалический барьер аминокислот, важных для синтеза нейротрансмиттеров.
Гомоцистеинурия — наследственное заболевание из группы аминоацидопатий, обусловленное нарушением метаболизма серосодержащих аминокислот, в частности, метионина. Дефицит цистатион-b-синтазы нарушает преобразование метионина в цистеин. Высокий уровень гомоцистеина связан с образованием некротически-дегенеративных участков в почках, селезенке, слизистой оболочке желудка и сосудах, активацией XII фактора свертывания, способствующего тромбообразованию.
Аргининосукциновая ацидурия вызывается мутациями в гене ASL, который кодирует фермент аргининосукцинатлиазу. Этот фермент катализирует превращение аргинино-янтарной кислоты в аргинин и фумарат на четвертом этапе цикла мочевины. Дефекты на этой стадии цикла мочевины приводят к накоплению в плазме аммиака, аргинино-янтарной кислоты, цитруллина и оротовой кислоты в моче, а также к дефициту аргинина в плазме. Ацидурия может иметь различную клиническую картину с началом в любом возрасте, включая период новорожденности. Состояние новорожденных обычно не вызывает подозрений в течение первых 24–48 часов после рождения, но в течение нескольких дней дебютирует тяжелая гипераммониемия, проявляющаяся летаргией, сонливостью, отказом от еды, рвотой, тахипноэ и респираторным алкалозом. Если не начать лечение, может произойти обострение летаргии, судороги, кома и смерть. Позднее начало ацидурии обычно индуцировано острой инфекцией, стрессом или высоким потреблением белка. Сообщалось также о поздних когнитивных дефектах или нарушениях обучаемости при отсутствии эпизодов гипераммониемии. У некоторых пациентов заболевание может протекать бессимптомно, несмотря на четкие биохимические признаки.
Тирозинемия 1 типа — заболевание, обусловленное дефицитом фумарилацетоацетатгидролазы, в результате чего происходит накопление высокотоксичных фумарил- и малеилацетоацетата, обладающих гепатотоксическим и канцерогенным действием. Конечные метаболиты — сукцинилацетон и сукцинилацетоацетат — являются митохондриальными токсинами, тормозящими фосфорилирование и блокирующими цикл Кребса. Накопление токсинов приводит к прогрессирующему заболеванию печени с развитием печеночной недостаточности, цирроза, тубулопатии с формированием ренальной тубулопатии, гипофосфатемического рахита, синдрома Фанкони. Острая тирозинемия сопровождается развитием гипертрофической кардиомиопатии. Кроме того, нарушается путь синтеза порфирина, ингибируется синтез порфобилиногена, что приводит к кризам, проявление которых напоминает порфирию. Все пациенты подвержены высокому риску развития гепатоцеллюлярной карциномы, вторичной по отношению к циррозу. Без своевременного лечения дети погибают в возрасте 10 лет.

Неонатальный скрининг проводят в родильном доме, однако некоторым детям по разным причинам его переносят на более поздние сроки, и тогда в амбулаторных условиях необходимо довести до родителей всю важность данного обследования и провести его.
Фенилкетонурия (ФКУ) – наследственное заболевание, в основе которого лежит нарушение обмена аминокислоты фенилаланина, которая, накапливаясь в крови и спинномозговой жидкости, вызывает поражение нервной системы. Частота ФКУ среди новорождённых 1 на 5000-10 000 (в России – 1 на 6950). Отставание в развитии ребёнка выявляется во втором полугодии жизни. Примерно у 60% больных отмечают идиотию, у остальных – менее выраженные умственные нарушения. Раннее выявление заболеваний у новорождённых, своевременное и правильное лечение таких больных с первых дней жизни предупреждает задержку умственного развития детей.
Врождённый гипотиреоз – одно из наиболее часто встречающихся заболеваний щитовидной железы вследствие нарушения синтеза её гормонов. Его частота составляет 1 случай на 4000-5000 новорождённых. В России ежегодно рождается 400 детей с врожденным гипотиреозом. В основе заболевания лежит полная или частичная недостаточность тиреоидных гормонов, приводящая к задержке развития всех органов и систем. В первую очередь страдает ЦНС и интеллект. При поздней диагностике и несвоевременном лечении дети становятся инвалидами с полной утратой способности к обучению, трудоспособности и к социальной адаптации. Своевременно начатое лечение тиреоидными гормонами предотвращает развитие умственной отсталости. Эффективность лечения зависит от срока постановки диагноза, так как уже в первые месяцы жизни наступают необратимые изменения в умственном развитии и росте скелета.
Неонатальный скрининг позволяет диагностировать гипотиреоз в первый месяц жизни ребёнка.
Адреногенитальный синдром – наследственное заболевание, обусловленное снижением активности фермента, участвующего в выработке гормонов надпочечника (кортизола и альдостерона). Распространённость, по данным разных авторов, колеблется от 1:5000 до 1:20 000 новорождённых. Клинические проявления зависят от того, на каком уровне блокируются ферменты. Наиболее тяжёлой, опасной для жизни является сольтеряющая форма, частота которой 1:27 000. Болезнь начинается в первую неделю жизни ребёнка, протекает остро, с выраженным обезвоживанием, падением артериального давления, судорогами и требует немедленного проведения реанимационных мероприятий с целью коррекции водно-электролитного баланса. При отсутствии адекватной терапии больные новорождённые умирают на 1-2-м месяце жизни. Для лечения назначают заместительную гормональную терапию (глюко- и минералкортикоиды).
Галактоземия – наследственное заболевание, связанное с невозможностью использования организмом углевода молока – галактозы. Частота болезни – 1 случай на 15 000-20 000 новорождённых. В основе её лежит отсутствие или резкое снижение активности ферментов, которые в процессе обмена веществ превращают галактозу молока в глюкозу. Вследствие неполного расщепления промежуточные продукты обмена оказывают токсическое воздействие на организм. Болезнь проявляется в виде тяжёлого поражения печени, нервной системы, глаз и других органов. Значительная часть больных, не получающих адекватной терапии, умирает в грудном возрасте; у других уже в первом полугодии жизни формируется тяжёлая инвалидизирующая патология: катаракта, цирроз печени, задержка нервнопсихического развития. При ранней постановке диагноза и своевременно начатом лечении сохраняется нормальный интеллект, не появляются нарушения глаз и печени. В настоящее время разработано и успешно применяется патогенетическое лечение диетой. Так как молоко (материнское и коровье) содержит лактозу, которая под действием ферментов расщепляется до галактозы и глюкозы, то с первых дней жизни, с момента установления диагноза, ребёнок должен быть переведён на безмолочное питание. Следует немедленно прекратить грудное вскармливание!
Технология проведения неонатального скрининга.
Проведение неонатального скрининга на наследственную патологию основано на определении дефектов ферментов, участвующих в обмене белков и углеводов, в сухом пятне крови на специальной фильтровальной бумаге (тест-бланке). Эти исследования проводят в медико-генетической консультации (центре), куда направляются образцы крови новорождённых одновременно на фенилкетонурию, врождённый гипотиреоз, адреногенитальный синдром, галактоземию, муковисцидоз.
Обязательным условием точности диагностики является тщательная пропитка кровью пятна на тест-бланке.
Забор образцов крови новорождённым в роддоме, в отделении выхаживания недоношенных или патологии новорождённых в детских больницах осуществляется специально подготовленной медсестрой. Кровь берут из пятки новорождённого через 3 ч после кормления: у доношенного ребёнка – на 4-й день жизни, у недоношенного – на 7-й день.
Алгоритм действий медсестры при взятии образцов крови:
– вымыть руки (гигиенический уровень), надеть перчатки;
– вымыть пятку ребёнка;
– протереть пятку стерильной салфеткой, смоченной 70% спиртом, промокнуть её сухой стерильной салфеткой;
– проколоть пятку стерильным одноразовым скарификатором;
– снять первую каплю крови стерильным сухим тампоном;
– мягко надавить на пятку для получения второй капли крови;
– приложить перпендикулярно тест-бланк к капле крови и пропитать его кровью насквозь;
– аналогичным образом нанести на тест-бланк 6-8 капель, вид пятен крови должен быть одинаковым с обеих сторон.
– высушить тест-бланк в горизонтальном положении на чистой обезжиренной поверхности не менее 2 ч без применения тепловой обработки и попадания прямых солнечных лучей;
– упаковать тест-бланки в чистый конверт таким образом, чтобы пятна крови не соприкасались.
Оформление сопроводительной документации. После забора образцов крови медсестра разборчиво записывает шариковой ручкой на тест-бланке, не затрагивая пятен крови, следующие сведения:
– наименование учреждения, в котором произведён забор образцов крови;
– фамилия, имя, отчество матери ребёнка;
– адрес выбытия матери ребёнка;
– порядковый номер тест-бланка с образцом крови;
– дата и номер истории родов;
– дата взятия образца крови;
– состояние ребёнка (здоров/болен);
– доношенный/недоношенный (срок гестации);
– масса тела ребёнка;
– фамилия, имя, отчество лица, осуществлявшего забор крови.
Источник: Сестринский уход за новорожденным в амбулаторно-поликлинических условиях.Под ред. Д.И. Зелинской. 2010г.
Читайте также: