
Наследственные липидозы патанатомия реферат
Обновлено: 04.07.2024
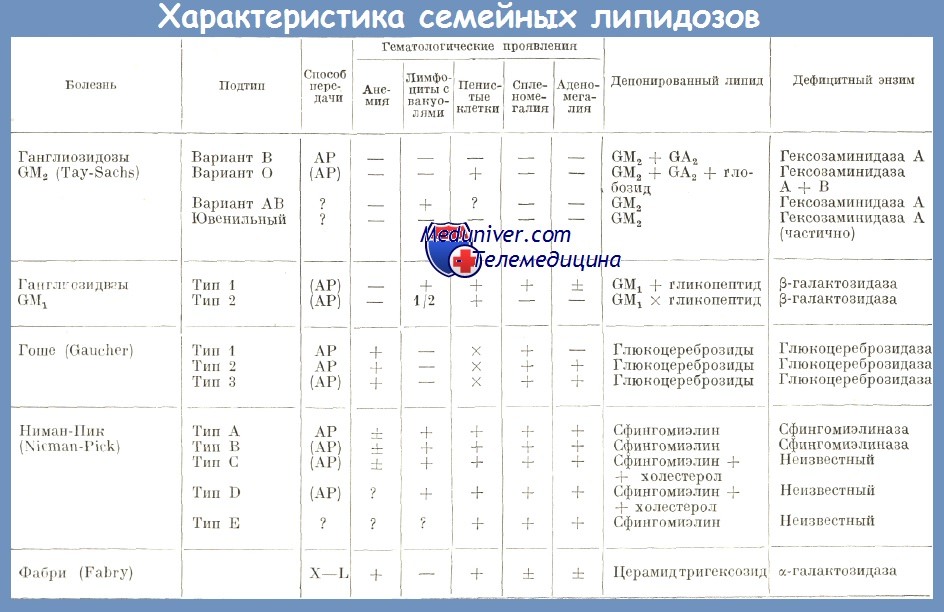
Группу наследственных липидозов составляют системные липидозы, возникающие вследствие наследственного дефицита ферментов, уч-щих в метаболизме определенных липидов, Поэтому системные липидозы относят к наследственным ферментопатиям (болезни накопления), т.к. дефицит фермента определяет накопление субстрата, т.е. липидов в клетках. В зависимости от вида накапливающихся в клетках липидов различают: 1) цереброзидлипидоз или глюкозилцерамидлипидоз – б-нь Гоше (возникают при дефиците фермента глюкоцереброзидаза; локализуются в печени, селезенке, костном мозге, ЦНС – у детей); 2) сфингомиелинлипидоз – б-нь Ниманна-Пика (возникает при дефиците фермента сфингомиелиназы; локализуются в печени, селезенке, костном мозге, ЦНС); 3) ганглиозидлипидоз – б-нь Тея-Сакса или амавротическая идиотия (возникает при дефиците фермента гексозаминидазы; локализуются в ЦНС, сетчатке глаз, нервных сплетениях, селезенке, печени); 4) генерализованный ганглиозидоз – б-нь Нормана-Лангинга (возникает при дефиците фермента β-галактозидазы; локализуются в ЦНС, нервных сплетениях, селезенке, печени, костном мозге, почках).
9. Стромально-сосудистые дистрофии, их деление на белковые (диспротеинозы), жировые (липидозы) и углеводные.
Стромально-сусудистые (мезенхимальные) дистрофии развиваются в рез-те нарушений обмена соед.тк. и выявляются в строме органа и в стенках сосуда. Они развиваются на территории гистиона, который образован отрезком микроциркуляторного русла с окружающими его элементами соед.тк. (основное в-во волокнистой структуры, клетки) и нервными волокнами. В связи с этим, среди мех-ма развития стромально-сосудистых дистрофий, преобладание нарушений транспортных систем трофики, общность морфогенеза, возможность сочетания не только различных видов дистрофии, но и перехода одного вида в другой. При нарушениях обмена соед.тк., преимущественно в ее межклеточном в-ве, накапливаются продукты метаболизма, которые могут приноситься с кровью и лимфой, быть рез-ом извращенного синтеза или появляться в рез-те дезорганизации основного в-ва и волокон соед.тк. В зависимости от вида нарушенного обмена мезенхимальные дистрофии делят на: 1) белковые (диапротеинозы); 2) жировые (липидозы); 3) углеводные.
10. Стромально-сосудистые белковые дистрофии, мукоидное набухание, фибриноидное набухание (фибриноид), гиалиноз. Морфологическая характеристика, причины, патогенез, исходы. Амилоидоз: классификация, морфогенез, патологическая анатомия; приобретённый (вторичный) амилоидоз, его причины.
К стромально-сосудистым диспротеинозам относят: 1) мукоидное набухание; 2) фибриноидное набухание; 3) гиалиноз; 4) амилоидоз. Мукоидное набухание – увеличение количества и перераспределение мукополисахаридов, преимущественно гликозаминогликанов (за счет отщепления их от белка), в основном веществе соединительной ткани. Накопление гликозаминогликанов всегда начинается с повреждения сосудов микроциркуляторного русла, что ведет к развитию тканевой гипоксии, активации гиалуронидазы и ослабеванию связи между гликозаминогликанами и белком. Гликозаминогликаны обладают выраженными гидрофильными свойствами, что ведет к выраженной гидратации (набуханию) основного вещества соединительной ткани. Микро-: коллагеновые волокна обычно сохраняют пучковое строение, но набухают и разволокняются. Набухание и увеличение в объеме основного вещества приводит к тому, что клетки соединительной ткани удаляются друг от друга. Макро-: органы практически не изменены. Локализация: в стенках артерий, сердечных клапанах, эндо- и эпикарде, в капсулах суставов. Причины: инфекционно-аллергические заболевания; ревматические болезни (ревматизм, системная красная волчанка, системная склеродермия, ревматоидный артрит, узелковый периартериит и др.); атеросклероз; гипертоническая болезнь; гипоксия. Исход: благоприятный - полное восстановление структуры и функции; неблагоприятный - может перейти в фибриноидное набухание. Фибриноидное набухание – глубокая и необратимая дезорганизация соединительной ткани, в основе которой лежит распад белка (коллагена, фибронектина, ламинина) и деполимеризация ГАГ, что ведет к деструкции ее основного вещества и волокон, сопровождающейся резким повышением сосудистой проницаемости и образованием фибриноида (- это сложное вещество, образованное за счет белков и полисахаридов, распадающихся коллагеновых волокон и основного вещества). Обязательным компонентом фибриноида является фибрин. Микро-: пучки коллагеновых волокон становятся гомогенными, эозинофильными, резко ШИК-позитивными, что свидетельствует о значительном увеличении в них количества гликопротеидов. Макро-: органы и ткани, в которых развивается фибриноидное набухание, мало изменены. Причины: инфекционно-аллергических заболеваниях (фибриноид сосудов при туберкулезе с гиперергическими реакциями); аллергических и аутоиммунных болезнях (ревматические болезни, гломерулонефрит); ангионевротических реакциях (фибриноид артериол при гипертонической болезни и артериальных гипертензиях); при хроническом воспалении. Исход: иногда развивается фибриноидный некроз, характеризующийся полной деструкцией соединительной ткани; вокруг очагов некроза обычно выражена реакция макрофагов; в дальнейшем происходит замещение очага деструкции рубцовой соединительной тканью (склероз) или гиалинозом. При гиалинозе, или гиалиновой дистрофии, в соединительной ткани образуются однородные полупрозрачные плотные массы (гиалин), напоминающие гиалиновый хрящ. Гиалин – это фибриллярный белок. Гиалиноз может развиваться в исходе разных процессов: плазматического пропитывания; фибриноидного набухания (фибриноида); склероза. Классификация. Различают: 1) гиалиноз сосудов; 2) гиалиноз собственно соединительной ткани. Каждый из двух видов гиалиноза может носить системный и местный характер. Гиалиноз сосудов - подвергаются преимущественно мелкие артерии и артериолы. Ему предшествуют повреждение эндотелия, базальной мембраны и гладкомышечных клеток стенки сосуда и пропитывание ее белками плазмы крови. Причины системного гиалиноза сосудов: гипертоническая болезнь; гипертонические состояния, гипертензии (болезни почек, опухоли эндокринных и половых желез); диабет (диабетический артериологиалиноз); ревматические заболевания; атеросклероз. В патогенезе ведущими механизмами его развития являются: деструкция волокнистых структур; повышение сосудисто-тканевой проницаемости (плазморрагия). С плазморрагией связаны пропитывание ткани белками плазмы и адсорбция их на измененных волокнистых структурах с последующей преципитацией и образованием белка – гиалина. Гиалиноз мелких артерий и артериол носит системный характер, но наиболее выражен в почках, головном мозге, сетчатке глаза, поджелудочной железе, коже. Микро-: артериолы превращаются в утолщенные стекловидные трубочки с резко суженным или полностью закрытым просветом. Исход: неблагоприятный, поскольку процесс необратим - гиалиноз мелких артерий и артериол ведет к атрофии, деформации и сморщиванию органа (например, развитие артериолосклеротического нефроцирроза). Гиалиноз собственно соединительной ткани - развивается обычно в исходе фибриноидного набухания, ведущего к деструкции коллагена и пропитыванию ткани белками плазмы и полисахаридами. Этот механизм развития системного гиалиноза соединительной ткани особенно часто встречается при заболеваниях с иммунными нарушениями (ревматические болезни). В основе гиалиноза в этих случаях лежат нарушения обмена соединительной ткани. Микро-: пучки коллагеновых волокон теряют фибриллярность и сливаются в однородную плотную хрящеподобную массу; клеточные элементы сдавливаются и подвергаются атрофии. Макро-: при выраженном гиалинозе волокнистая соединительная ткань становится плотной, хрящевидной, белесоватой, полупрозрачной. Исход. В большинстве случаев неблагоприятный в связи с необратимостью процесса, но возможно и рассасывание гиалиновых масс. Так, гиалин в рубцах – так называемых келоидах – может подвергаться разрыхлению и рассасыванию. Обратим гиалиноз молочной железы, причем рассасывание гиалиновых масс происходит в условиях гиперфункции желез. Иногда гиалинизированная ткань ослизняется. Амилоидоз – это стромально-сосудистый диспротеиноз, который сопровождается глубоким нарушением белкового обмена и появлением аномального фибриллярного ультраструктурно, но светооптически гомогенного белка с отложением его в межуточной ткани и стенках сосудов. Клиническая классификация амилоидоза основана на типе белка и типе ткани, в которой он накапливается, распространенности и возможной причине его возникновения: 1) системный амилоидоз: первичный системный амилоидоз с преимущественным накоплением амилоида в сердце, желудочно-кишечном тракте, языке, коже и нервах. Эта локализация отмечается при первичном амилоидозе и при новообразованиях из B-лимфоцитов. Вторичный амилоидоз с преимущественным накоплением амилоида в печени, селезенке, почках, кишечнике, надпочечниках. Он возникает при хронических воспалительных заболеваниях, таких как туберкулез, сифилис, лепра, хронические нагноительные процессы (остеомиелит, бронхоэктатическая болезнь, хронический пиелонефрит). 2) ограниченный (местный) амилоидоз: ограниченный амилоидоз может иметь узловую, опухолеподобную форму. Он встречается редко и наблюдается в языке, мочевом пузыре, легких и коже. Накопление амилоида обычно связано с небольшими новообразованиями из плазматических клеток. При болезни Альцгеймера, скопления амилоида особого типа определяются во внеклеточном веществе мозга (в виде бляшек). 3) амилоид в новообразованиях: амилоид накапливается в строме большого количества эндокринных новообразований, например, медуллярного рака щитовидной железы. 4) семейный врожденный амилоидоз: семейный амилоидоз был описан в небольшом количестве семей. 5) семейный амилоидоз классифицируется на нейропатический, нефропатический и сердечный, в зависимости от преобладания поражения той или иной системы. 6) сенильный амилоидоз: небольшие количества амилоида (АS) часто обнаруживаются в сердце, поджелудочной железе и селезенке у пожилых людей. Это особый тип амилоида, составленного из полипептидов, синтезируемых островковыми клетками, которые имеют гормональную активность, воздействуя на утилизацию глюкозы в мышцах. Макро- и микроскопия амилоидоза. Внешний вид органов при амилоидозе зависит от степени развития процесса. Если отложения амилоида небольшие, внешний вид органа изменяется мало и амилоидоз диагностируется лишь при микроскопическом исследовании. При выраженном амилоидозе органы увеличиваются в объеме, бледные, с сальным блеском Поврежденные ткани имеют более плотную консистенцию и сниженную эластичность по сравнению с нормальными тканями. Признаками наиболее выраженного поражения ткани являются бледно-серый оттенок и своеобразный восковидный, или сальный, вид ее на разрезе. В селезенке амилоид может откладываться как изолированно в лимфатических фолликулах (амилоидно измененные фолликулы увеличенной и плотной селезенки на разрезе имеют вид полупрозрачных зерен, напоминающих зерна саго - саговая селезенка), так и равномерно по всей пульпе (селезенка увеличена, плотная, коричнево-красная, гладкая, имеет сальный блеск на разрезе сальная селезенка). В почках амилоид откладывается в стенках приносящих и выносящих артериол, в капиллярных петлях и мезангии клубочков, в базальных мембранах канальцев и в строме. Почки становятся плотными, большими и "сальными". По мере нарастания процесса развивается амилоидное сморщивание почек. В печени отложение амилоида наблюдается между звездчатыми ретикулоэндотелиоцитами синусоидов, по ходу ретикулярной стромы долек, в стенках сосудов, протоков и в соединительной ткани портальных трактов. По мере накопления амилоида печеночные клетки атрофируются и погибают. При этом печень увеличена, плотная, выглядит "сальной". В кишечнике амилоид выпадает в строме ворсин слизистой оболочки, а также в стенках сосудов как слизистой оболочки, так и подслизистого слоя. При резко выраженном амилоидозе железистый аппарат кишечника атрофируется. Амилоидоз надпочечников - двусторонний, отложение амилоида встречается в корковом веществе по ходу сосудов и капилляров. В сердце амилоид обнаруживается под эндокардом, в волокнах и сосудах стромы, а также в эпикарде по ходу вен --- резкое его увеличению (амилоидная кардиомегалия). Оно становится очень плотным, миокард приобретает сальный вид. В скелетных мышцах, как и в миокарде, амилоид выпадает по ходу межмышечной соединительной ткани, в стенках сосудов и в нервах. Периваскулярно и периневрально нередко образуются массивные отложения амилоидного вещества. Мышцы становятся плотными, полупрозрачными. В легких отложения амилоида появляются сначала в стенках разветвлений легочных артерии и вены, а также в перибронхиальной соединительной ткани. Позже амилоид появляется в межальвеолярных перегородках. В головном мозге при старческом амилоидозе амилоид находят в сенильных бляшках коры, сосудах и оболочках. В коже - хар-ется диффузным отложением амилоида в сосочках кожи и ее ретикулярном слое, в стенках сосудов и базальных мембранах сальных и потовых желез, что сопровождается деструкцией эластических волокон, резкой атрофией эпидермиса и придатков кожи. В поджелудочной железе - помимо поражения артерий железы, встречается и амилоидоз островков, что наблюдается в глубокой старости. Исход: неблагоприятный, практически необратимый. Функциональное значение определяется степенью развития амилоидоза. Выраженный амилоидоз ведет к дистрофии и атрофии паренхимы и склерозу стромы органов, к их функциональной недостаточности. При выраженном амилоидозе чаще всего наблюдается хроническая почечная, реже – печеночная, сердечная, легочная, надпочечниковая, кишечная (синдром нарушенного всасывания) недостаточность.
11. Стромально-сосудистые жировые дистрофии, связанные с нарушением обмена нейтрального жира, холестерина и его эстеров. Общее ожирение (тучность), причины, патогенез, морфологическая характеристика, классификация. Истощение: причины, патогенез, морфологические проявления. Атеросклероз, как пример заболевания нарушенного обмена холестерина и его эстеров.
Стромально-сосудистые жировые дистрофии возникают при нарушениях обмена лабильного жира (нейтральных жиров) или холестерина и его эфиров. Нейтральные жиры – это лабильные жиры, обеспечивающие энергетические запасы организма. В свободном состоянии они локализуются в жировых клетках жировых депо: подкожной, забрюшинной клетчатки и клетчатки средостения, брыжейки, сальника, эпикарда, костного мозга. Жировая ткань выполняет не только обменную, но и опорную, механическую функцию, поэтому она способна замещать атрофирующиеся ткани. Нарушение обмена нейтральных жиров проявляется в увеличении их запасов в жировой ткани. Оно может быть общим и местным. Ожирение, или тучность,– увеличение количества нейтральных жиров в жировых депо. Оно носит общий характер и выражается в избыточном отложении жиров в подкожной клетчатке, сальнике, брыжейке кишечника, средостении, эпикарде. Различают: 1) первичное (идиопатическое) ожирение – его причина неизвестна; 2) вторичное ожирение. Виды вторичного ожирения: алиментарное (несбалансированное питание и гиподинамия); церебральное (при опухолях мозга, особенно гипоталамуса, некоторых нейротропных инфекциях); эндокринное (синдром Иценко-Кушинга, адипозо-генитальная дистрофия, гипотиреоз, гипогонадизм); наследственное (болезнь Гирке). По внешним проявлениям различают универсальный симметричный тип ожирения, который делят на три подтипа: 1) верхний (хар-ется накоплением жира преимущественно в области подкожной клетчатки лица, затылка, шеи, верхнего плечевого пояса, молочных желез); 2) средний (сопровождается отложением жира в подкожной клетчатке живота в виде фартука); 3) нижний (избыток жировой клетчатки наблюдается в области бедер и голеней). По превышению массы тела больного выделяют четыре степени ожирения: I степень ожирения – избыточная масса тела составляет до 30%; II степень ожирения – избыточная масса тела составляет до 50%; III степень ожирения – избыточная масса тела составляет до 99%; IV степень ожирения – избыточная масса тела составляет от 100% и более. По числу и размеру адипозоцитов выделяют два варианта: 1) гипертрофический; 2) гиперпластический. При гипертрофическом варианте ожирения число адипозоцитов не изменяется, но жировые клетки многократно увеличены в объеме за счет избыточного содержания в них триглицеридов. Клиническое течение заболевания злокачественное. При гиперпластическом варианте число адипозоцитов увеличено. Однако, метаболические изменения в них отсутствуют. Течение болезни доброкачественное. При тучности большое клиническое значение имеет ожирение сердца. Жировая ткань разрастается под эпикардом и прорастает между мышечными пучками, сдавливая их и охватывая сердце в виде футляра. Это ведет к атрофии мышечных волокон. Обычно ожирение резко выражено в правой половине сердца, что приводит к замещению миокарда жировой тканью, в связи с чем может произойти разрыв сердца. Исход общего ожирения редко бывает благоприятным. Антиподом общего ожирения является истощение, в основе которого лежит общая атрофия. Местное увеличение количества жировой клетчатки обозначается термином липоматоз. Примером липоматоза может служить болезнь Деркума (lipomatosis dolorosa). Она характеризуется появлением в подкожной клетчатке конечностей и туловища узловатых болезненных отложений жира, напоминающих по внешнему виду опухоль (липому). Причиной этого заболевания является полигландулярная эндокринопатия. Примером липоматоза может служить также вакатное ожирение (жировое замещение) ткани или органа при атрофии (жировое замещение почки или вилочковой железы при их атрофии).
Липидозы (lipidoses; липид [ы] + -ōsis) — группа заболеваний, характеризующихся нарушением липидного обмена и имеющих преимущественно наследственный характер. Большинство Липидозов. относится к болезням накопления, которые обусловлены отложением аномально больших количеств нерасщепленных продуктов жирового обмена в различных органах и тканях, что приводит к значительному нарушению их функции.
Прикрепленные файлы: 1 файл
Lipidozy.ppt
Карагандинский государственный медицинский университет
Кафедра патологической анатомии
На тему: Липидозы
Выполнил: ст-нт 258 гр. ОМФ
- Липидозы (lipidoses; липид [ы] + -ōsis) — группа заболеваний, характеризующихся нарушением л ипидного обмена и имеющих преи мущественно наследственный хар актер. Большинство Липидозов. относится к болезням накоплени я, которые обусловлены отложением аномально больших количеств н ерасщепленных продуктов жирово го обмена в различных органах и тканях, что приводит к значительному н арушению их функции. В основе Липидозов. лежит полная или частичная нед остаточность лизосомальных фер ментов, участвующих в обмене липидов и обусловленная наследственным дефектом соответствующего гена . Большинство Липидозов. наследуется по аутосомно-рецес сивному типу, исключение составляет болезнь Фабри, которая наследуется по Х-сцепл енному, рецессивному типу.
- К липидозам относятся ганглиоз идозы — GM1-ганглиозидоз (тип I, II, III), GM2-ганглиозидоз (тип I, II, ювенильный, хронический тип); болезнь Ниманна — Пика; болезнь Гоше; болезнь Фабри; болезнь Краббе; болезнь I-клеток.
- группа заболеваний, обусловленных нарушением катаб олизма ганглиозидов — сложных гликолипидов, углеводная часть молекулы кото рых содержит остаток сиаловой кислоты. Выделяют несколько типов ОМ, и ОМ; ганглиозидозов. В основе их развития лежит мет аболический дефект расщепления ганглиозидов в результате нед остаточности специфических лиз осомальных гидролаз.
- На вскрытии обнаруживают расширение желудочков мозга и его атрофию в результате гибели нейронов. Гистологически выявляются пенистые гистиоциты в костном мозге, печени, селезенке, лимфатических узлах и др.
Мазок крови при ганглиозидозе
болезнь Ниманна — Пика
- При болезни Ниманна — Пика сфи нгомиелин в избыточном количес тве накапливается в этих клетк ах или их компонентах. Сфингомиелин обнаруживают в пе чени, селезенке, лимфатических узлах, альвеолах и миокарде. Клинически болезнь Ниманна — П ика проявляется гепатоспленоме галией с рождения, прогрессирующим поражением нер вной системы и снижением интел лекта.
- Головной мозг макроскопически не изменен. При микроскопическом исследовании выявляются суданофильные клетки накопления (клетки Пика), в большом количестве содержащиеся во многих органах как среди клеток макрофагально-гистиоцитарной системы, так и в эпителии
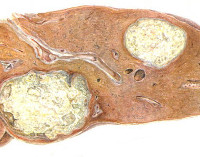
- Селезенка значительно увеличена, плотная, кирпично-красного цвета, на разрезе пестрая из-за чередования кирпично-красных и желтоватых участков. Лимфатические узлы увеличены, на разрезе цвета яичного желтка. Печень значительно увеличена, уплотнена, от охряно-желтого до канареечного цвета, на разрезе имеет глинистый вид. Надпочечники увеличены, светлее, чем в норме. В легких отмечаются мелкие очаги, напоминающие милиарные туберкулы, или сетчатая инфильтрация желтоватого цвета. Почки менее увеличены, корковое вещество их очень бледное
- (глюкоцереброзидный липидоз, г люкоцереброзидоз) — одно из на иболее частых наследственных н арушений гликолипидного обмена . Предполагают, что в основе б олезни Гоше лежат аллельные му тации генов, кодирующих глюкоц ереброзидазу — фермент, катали зирующий гидролитическое отщеп ление глюкозы от глюкоцереброз ида, который накапливается в р азличных органах и тканях.
- Источником накапливающегося глюкоцереброзида являются лейкоциты и эритроциты. Чрезмерное увеличение печени и селезенки объясняют помимо накопления глюкоцереброзида компенсаторной реакцией клеток с целью получения дополнительного количества глюкоцереброзидазы. Наполненные глюкоцереброзидом клетки с эксцентрически смещенным ядром и цитоплазмой, напоминающей смятую папиросную бумагу или безе, носят название клеток Гоше. Их обнаруживают в лимфатических узлах, капиллярах альвеол, костном мозге, в адвентиции артериол, вен, лимфатических сосудов. В гистопатологической картине ц.н.с. преобладает гибель нейронов, клетки Гоше в зоне поражения единичны.
- (диффузная универсальная ангио кератома, наследственный дисто нический липидоз) — врожденный дефект катаболизма гликосфинг олипидов с преобладанием пораж ения почек.
- Энзимный дефект приводит к системному отложению сфингогликолипидов и гликопротеида в пораженных тканях, в частности в эндотелии и гладкой мускулатуре сосудов, сердце, почках (почечные клубочки и канальцы), глазах (эпителиальные клетки роговицы), ганглиях вегетативной нервной системы.
- Болезнь Краббе (глобоидно-клеточная лейкодист рофия) — быстро прогрессирующее демие линизирующее дегенеративное за болевание ц.н.с. Тип наследования — аутосомно-р ецессивный. В основе болезни лежит снижени е активности фермента галактоз илцерамид-b-галактозидазы, который в норме расщепляет гал актоцереброзид до церамида и г алактозы. В головном мозге, печени, селезенке, почках, лейкоцитах, фибробластах накапливаются гал актоцереброзид и его производн ое психозин. Количество последнего повышает ся в 10—100 раз, что оказывает токсическое возд ействие на олигодендроглиальны е клетки, формирующие миелиновую оболочк у. В зонах демиелинизации вокруг мелких кровеносных сосудов бел ое вещество содержит большое к оличество глобоидных гистиоцит ов (макрофагов). Уменьшение олигодендроглиальны х клеток сопровождается глиозо м. Периферические нервы подвергаю тся аксональной дегенерации с накоплением пенистых гистиоцит ов.
- заболевание, которое связано с нарушением р асщепления мукополисахаридов и сфингогликолипидов. В связи с этим клинически оно проявляется симптомами, свойственными мукополисахаридо зам и болезни Ниманна-Пика (спланхномегалией, грубыми чертами лица, задержкой умственного развития и неврологическими расстройст вами).
- В фибробластах больных с болезнью I-клеток отмечается дефицит b-гексозаминидазы, арилсульфатазы А и b-глюкоронидазы, в жидких средах организма (сыворотке крови, моче, цереброспинальной жидкости) активность лизосомных гидролаз повышена. Дефект маркерного центра лизосомных ферментов обусловлен недостаточностью фосфотрансферазы, локализованной в пластинчатом комплексе аппарата Гольджи. При электронной микроскопии фибробластов определяются необычные цитоплазматические включения. Такие же изменения отмечаются в аксонах периферических нервов, печени, почечных клубочках и эпителии канальцев почек. При гистологическом исследовании мозга больного обнаруживаются увеличенные в размерах лизосомы, содержащие гранулярные массы диаметром от 5 до 15 ммк, окрашивающиеся метахроматически. При прижизненном исследовании мозга с помощью метода магнитно-резонанс ной томографии обнаруживаются очаги демиелинизации, наиболее часто концентрирующиеся вокруг четвертого желудочка и субкортикально.
Липоидозы – наследственные заболевания, связанные с нарушением метаболизма жиров, отложением липидов и их метаболитов в различных органах и тканях. Общие клинические проявления представлены прогрессирующим расстройством интеллектуальных и двигательных функций, поражением костей, кожи, центральной нервной системы, глаз и внутренних органов (печени, почек, селезенки). Диагностика основана на лабораторных исследованиях ферментативной активности, количества токсичного субстрата, наличия мутации в генах. Лечение включает ферментозаместительную, субстратредуцирующую и симптоматическую терапию.
МКБ-10
Общие сведения
Синонимичные названия липоидозов – липидозы, ретикулоэндотелиозы, лизосомные болезни накопления липидов. К данной группе относится сфингомиелиноз, болезнь Тея-Сакса, болезнь Гоше, семейные гиперлипидемии и некоторые другие заболевания. Общей характеристикой является патологическое накопление липидов внутри клеток организма в результате дефекта ферментных систем. Липоидозы относятся к группе редких (орфанных) заболеваний. Их распространенность очень низка, для отдельных типов патологий составляет от 1:40 тыс. до 1:1 млн. и реже. Суммарная частота – 1:7 тыс. Большинство липидозов имеют прогредиентное течение, приводят к инвалидизации и ранней смерти.
Причины липоидозов
Определяющим фактором развития липидозов является генетический дефект, который обуславливает полную или частичную недостаточность лизосомальных ферментов, расщепляющих сложные липиды. Наследование болезней происходит по аутосомно-рецессивному механизму. Это означает, что новорожденный оказывается больным, если получает мутационный ген от каждого родителя (имеет пару дефектных генов в аллели). Когда мутация передается только от матери или отца, то ребенок является ее носителем и остается здоровым.
Исключительный механизм передачи дефекта у болезни Фабри. В отличие от других липидозов, она наследуется по X-сцепленному рецессивному типу. Гемизиготные пациенты мужского пола больны, передают мутацию только дочерям. У девочек заболевание всегда проявляется при наличии двух рецессивных (измененных) генов. Иногда симптомы липидоза проявляются у пациенток с одним дефектным геном, когда доминантный ген оказывается инактивированным (причины этого не выяснены).
Патогенез
Патогенетической основой большинства липоидозов является генетически детерминированный ферментативный дефект – энзимопатия. Вследствие этого лизосомы клеток накапливают липиды и промежуточные продукты их метаболизма, что ведет к прогрессивному нарастанию нарушения функций органов. При синдроме Вольмана определяется недостаточность кислой этеразы, становится невозможным полный цикл обмена холестерола, повышается содержание его эфиров в лизосомах селезенки, печени, надпочечников, кишечника и костного мозга. У пациентов с болезнью Гоше снижено или полностью отсутствует производство бета-глюкозидазы; в печени, селезенке, костном мозге скапливаются продукты расщепления сфинголипидов. Болезнь Нимана-Пика развивается на базе дефицита сфингомиелазы, характеризуется повышением количества сфингомиелина во многих клетках, особенно в гепатоцитах, нейронах. При идиотии Тэя-Сакса существует дефект N-ацетилгексозаминидазы, происходит накапливание ганглиозидов в головном мозге.
Классификация
Наследственные нарушения метаболизма сложных липидов представлены группой различных по происхождению заболеваний, которые на патогенетическом уровне связаны с патологией жирового обмена. Для липоидозов характерно скопление сложных липидных соединений внутри лизосом клеток. В зависимости от того, какой липид не расщепляется до конца и откладывается в тканях, выделяют несколько типов болезней:
- Гликолипидозы. При данном типе заболеваний невозможен полный распад гликолипидов – соединений, состоящих из углеводов и жирных кислот. Гликолипидозы представлены цереброзидозами (сфинголипидоз Гоше, болезнь Краббе), сульфатидозами (метахромотическая лейкодистрофия), церамидолигозидозами (болезнь Фабри, церамидлактозидлипоидоз), ганглиозидозами (болезнь Сандхоффа, ранняя детская амавротическая идиотия).
- Липопротеинемии. Обусловлены патологией обмена липидов плазмы крови, основанной на генетическом дефекте ферментов или рецепторов клеток. Липиды плазмы – жирные кислоты, триглицериды, холестерин. Липопротеинемии включают семейную гиперхолестеринемию, комбинированную семейную гиперлипидемию и семейные гиперлипидемии типов I-V.
- Сфингомиелиноз. Синонимичное название – болезнь Ниманна-Пика. При развитии этого заболевания в ретикулоэндотелиальных клетках увеличивается содержание фосфолипидного соединения сфингомиелина.
Симптомы липоидозов
Клиническая картина липидозов определяется особенностями вовлечения в патологический процесс органов и систем. При болезни Гоше I типа поражается печень, селезенка, кости и костный мозг. Симптомы включают увеличение размеров печени, хрупкость костей, анемию, лейкопению, снижение свертываемости крови. II тип заболевания разворачивается с преимущественным повреждением ЦНС и печени. Наблюдаются судорожные приступы, мышечный гипертонус, спастичность, интеллектуальная недостаточность, расстройства акта глотания. У людей с галактозилцерамидным липидозом (болезнью Краббе) снижается функциональность миелиновой оболочки. Развивается гипервозбудимость, рвота, судороги, задержка психомоторного развития с прогрессирующим снижением интеллекта и зрения.
При метахромотической лейкодистрофии патологически изменяется миелиновая оболочка. Основные симптомы – гипотония мышц рук и ног, снижение (отсутствие) сухожильных рефлексов, атаксия, атрофия зрительных нервов, нистагм, спастический тетрапарез, глухота, интеллектуальное и моторное недоразвитие. Клинические признаки болезни Андерсона-Фабри разнообразны, одними из наиболее распространенных являются нейропатическая боль (умеренной и легкой выраженности, в ступнях и ладонях) и кризы Фабри – сильные жгучие боли в конечностях приступообразного характера. Дополнительно возникает гипогидроз или ангидроз, непереносимость физических нагрузок, ангиокератомы, расстройства слуха, сердечно-сосудистой и почечной функций.
При развитии синдрома Сандхоффа определяется общая вялость, гипотонус мышц конечностей, трудности сосания и глотания, прогрессирующая задержка моторного и психического развития, двигательная слабость, шумы в сердце, судороги, слепота, увеличение селезенки. При ранней детской амавротической идиотии больше всего поражается центральная нервная система. К концу первого полугодия ухудшается реакция детей на внешние сигналы (лица близких, игрушки), утрачиваются двигательные навыки, снижается познавательный и игровой интерес. Нарушается зрение вплоть до слепоты. Формируются судорожные припадки.
Первые симптомы сфингомиелиноза – вялость, малоподвижность, апатичность, отказ от еды, рвота. Позже увеличивается живот (гепатомегалия), конечности становятся худыми, кожа приобретает коричневатый оттенок, периоды заторможенности сменяются гипервозбуждением. Дети отстают в психофизическом развитии. Диагностируется умеренная гидроцефалия, гипертермия, спастический парез ног и рук, приступообразная асфиксия.
Проявления гиперлипопротеинемий обнаруживаются в возрасте до 10 лет. Для всех форм болезней свойственно отложение подкожного жира в виде ксантом, а также абдоминальные боли, панкреатит, гепатоспленомегалия (скопление липидов в селезенке, печени). При комбинированной семейной гиперлипидемии и гиперлипидемии II типа возможен ранний атеросклероз сосудов. При гиперлипидемиях IV и V типа – снижение чувствительности к глюкозе, ИБС.
Осложнения
Липоидозы протекают с развитием недостаточности жизненно важных органов. Наиболее частыми осложнениями становятся заболевания сердца и сосудов, ЦНС, почек, легких, печени. Пациенты страдают от атеросклероза сосудов, сердечной и дыхательной недостаточности, ишемической болезни сердца, хронической почечной и печеночной недостаточности, аденом и цирроза печени, инсультов, транзиторных ишемических атак. При типах заболеваний, совместимых с жизнью, зачастую нарушено двигательное и психическое развитие. Многие больные оказываются неспособны к самообслуживанию, обучению и овладению профессией, нуждаются в пожизненном уходе со стороны окружающих.
Диагностика
Липоидозы не являются узкоспециализированными заболеваниями, поэтому их выявлением и лечением занимаются педиатры, гематологи, гастроэнтерологи, ревматологи, неврологи, психиатры и врачи-генетики. На первичном этапе диагностики проводится сбор семейного анамнеза: при наследственном характере ферментопатии у больного могут быть родственники с подтвержденным диагнозом липидоза. При сборе клинических данных специалисты обращают внимание на время начала симптомов: чаще всего болезнь проявляется в период новорожденности или первого года жизни, редко – у детей постарше или взрослых. К специфическим методам обследования пациентов относят:
- Анализ активности дефектного фермента. Исследованию подвергаются различные биоматериалы – плазма крови, лейкоциты, сухие пятна крови, культура фибробластов кожи, биопсийный материал почек, печени. При липоидозах определяется недостаток активности определенного фермента: от легкого снижения до полного отсутствия.
- Количественное исследование липидов. В крови и биопсийном материале органов анализируется содержание патологически накапливаемых липидов и промежуточных продуктов их обмена. У больных липоидозом показатели превышают норму. Параллельно изучаются изменения строения пораженных клеток.
- Секвенирование ДНК. Выявление дефектного гена в хромосоме является наиболее точным, но трудоемким методом диагностики наследственных болезней. Широко применяется в рамках перинатальной и преимплантационной диагностики, в случаях, когда вышеперечисленные анализы не дают однозначного представления о диагнозе.
- Визуализирующие исследования органов. Дополнительно проводится диагностика состояния пораженных органов – сердца, печени, желчного пузыря, селезенки, легких, почек, головного мозга. Используется УЗИ, МРТ, КТ, ЭКГ, ЭЭГ. Процедуры позволяют оценить размеры, выявить структурные изменения и новообразования в органе.
Лечение липоидозов
Терапия данной группы заболеваний – сложная задача для врачей разных специальностей. Методы лечения несовершенны и продолжают разрабатываться, при некоторых видах болезней возможно добиться лишь временного улучшения самочувствия больного, при других достижима стойкая ремиссия. Общая схема медицинской помощи больным состоит из трех компонентов:
- Ферментозаместительная терапия. В организм пациентов вводятся препараты с искусственно выделенным дефицитарным ферментом. Инъекции выполняются пожизненно, позволяют восстановить метаболизм липидов.
- Субстратредуцирующая терапия. Лечение направлено на снижение интенсивности образования патологически накапливаемого соединения (сложного липида, его метаболитов). Используются молекулы с низкой массой, которые стимулируют остаточную активность фермента.
- Симптоматическая терапия. Препараты подбираются индивидуально на основании клинической картины болезни. Распространено применение антиконвульсантов, обезболивающих средств, ингибиторов АПФ, гепатопротекторов. При отдельных видах липоидозов использование симптоматических лекарств является единственным способом лечения.
Прогноз и профилактика
Липоидозы характеризуются гетерогенностью генетического дефекта и выраженной клинической полиморфностью. Большинство из них имеет непрерывное прогрессирующее течение. Правильная диагностика и своевременное лечение позволяют увеличить продолжительность жизни больных, а при легких формах способствуют улучшению адаптации. Профилактика возможна на этапе планирования и в первые месяцы беременности. Супружеским парам с высоким риском передачи болезни ребенку рекомендуется медико-генетическое консультирование, а в первом триместре – исследование амниотической жидкости и материала биопсии хориона на наличие мутаций генов.
1. Лизосомные болезни накопления липидов у детей/ Захарова И.Н. и др.// Медицинский совет. - 2016 - №1.
Определение понятия и сущности жировых дистрофий. Рассмотрение функции липидов в организме. Изучение липидоза, ожирения и истощения. Клиническая картина жировой дистрофии сердца, печени и почек. Ознакомление с проявлениями болезней Гоше и Намана-Пика.
| Рубрика | Медицина |
| Вид | презентация |
| Язык | русский |
| Дата добавления | 18.05.2014 |
| Размер файла | 669,7 K |
Студенты, аспиранты, молодые ученые, использующие базу знаний в своей учебе и работе, будут вам очень благодарны.
HTML-версии работы пока нет.
Cкачать архив работы можно перейдя по ссылке, которая находятся ниже.
Подобные документы
Общее понятие о липидозе, причины возникновения. Приобретенные паренхиматозные липидозы. Жировая дистрофия печени, почек и миокарда. Три класса сфинголипидов: сфингомиелины, ганглиозиды и цереброзиды. Болезнь Нимана—Пика, ювенильный ганглиозидоз.
презентация [2,4 M], добавлен 11.12.2014
Жировые дистрофии органов. Жировая дистрофия миокарда, печени, почек. Нарушение липидного метаболизма. Аутосомально-рецессивное наследование. Болезнь Ниманна—Пика, липогранулематоз Фарбера, болезнь Вулмена, псевдогурлевская полидистрофия, болезнь Краббе.
презентация [4,1 M], добавлен 29.05.2014
Накопление липидов в лизосомах. Мутация гена, контролирующего синтез фермента 7-d-глюкоцереброзидазы. Нарушение функции макрофагов. Основные типы болезни Гоше. Клиническая картина ненейронопатического типа и нейронопатической инфантильной формы.
презентация [3,8 M], добавлен 08.03.2016
Определение болезни Гоше и ее основная характеристика. Изучение причин возникновения данного заболевания. Клиническая картина и симптомы. Дифференциальная диагностика. Исследование методов лечения злокачественной и доброкачественной форм болезни Гоше.
реферат [179,2 K], добавлен 15.09.2014
Причины, способные вызвать повреждение, его характер и степень. Понятие и морфологическая сущность дистрофий, непосредственные причины их развития. Механизм повреждений и общая характеристика паренхиматозной, гидропической, роговой, жировой дистрофии.
лекция [41,6 K], добавлен 24.05.2009
Изучение классификации пищевых отравлений по К.С. Петровскому. Описания дистрофии, ожирения и остеопороза. Исследование симптомов токсикоза, авитаминоза, гиповитаминоза, алкогольного отравления. Клиническая картина и профилактика пищевого ботулизма.
презентация [353,3 K], добавлен 26.03.2013
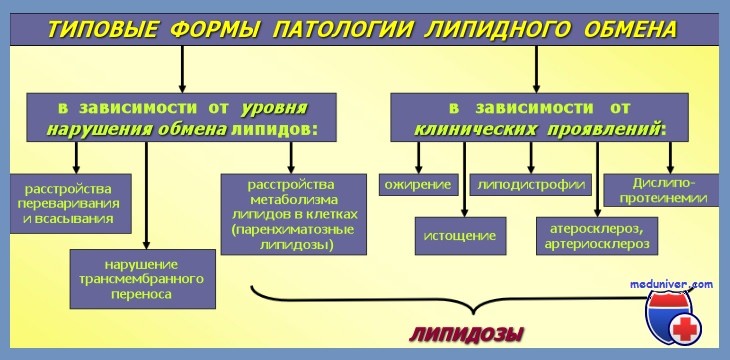
Причины, клиническая характеристика, диагностика и лечение нарушений липидного обмена. Ожирение, истощение, дислипопротеинемии, липодистрофии и липидозы. Жировая дистрофия, сопровождающаяся избыточным накоплением липидов в паренхиматозных клетках.
Липидозы — заболевания, характеризующиеся аномалийным накоплением сфинголипидов в макрофагах костного мозга и других тканей. Эти болезни известны в патологии под различными названиями: тезаурисмозы, гистиомоноцитарные липидозы, метаболические или аккумулятивные ретикулезы.
Патогенез липидозов. Главное расстройство, стоящее в основе всех липидозов, состоит в неспособности деградации сфинголипидов и их накоплении в макрофагах различных тканей.
Главный компонент различных сфинголипидов — церамид, который является ацилированным сфингозином. Она выполняет важнейшие структурные функции на уровне многих клеток в организме. Жирнокислотная часть сфинголипидов на уровне мозга представлена в особенности стеариновой кислотой (С18), а на уровне остальных тканей жирными кислотами с высоким числом углеродов (С20—С21). Отличительная черта и функция каждого сфинголипида определяются радикалом, с которым связан церамид.
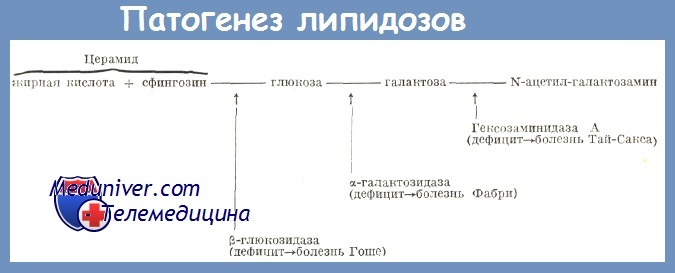
Большинство этих болезней возникают из-за врожденных энзиматических дефицитов: наследственные липидозы. При этом типе заболеваний накопление сфинголипидов происходит не из-за сверхпроизводства липидов, а из-за отсутствия или сокращения некоторых специфических гидролитических энзим (Brady, Turpin).
В других случаях не существует энзиматического дефицита, а накопление липидов происходит благодаря чрезмерному разрушению клеток (гранулоцитов, эритроцитов, тромбоцитов), которые высвобождают большие количества липидного материала — приобретенные или вторичные липидозы (Albrecht, Dosik, Kattlowe, Silverstein, Ursea, Zaino).
Независимо от механизма тезауризации, наиболее затронутыми тканями оказываются те, которые представляют повышенный турновер липидов: развивающаяся нервная система и моноцитомакрофаговая система, где, в результате фагоцитоза, катаболизм липидов составляет главную функцию этих клеток.

Изучение липидозов имеет значение для гематологов из следующих соображений:
а) некоторые болезни представляют гематологические явления (болезнь Гоше, Нимана-Пика); другие представляют кожные поражения петехиального типа (болезнь Фабри);
б) липиды, накопляющиеся в макрофагах, происходят от эритроцитов, гранулоцитов и тромбоцитов;
в) в течение гематологических заболеваний могут появляться вторичные липидозы.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Читайте также: