
Молекулярное моделирование биоструктур реферат
Обновлено: 04.05.2024
Таким образом, создание таких технологий не просто ускоряет или удешевляет исследования, а зачастую позволяет в принципе найти хоть что-нибудь от важнейших заболеваний. Таким путем, например, совсем недавно были получены молекулы от вирусного гепатита. Впервые в истории человечества вирусный гепатит С стал если не излечимым, то контролируемым заболеванием. Для определенного вида рака груди… Читать ещё >
Заключение. Молекулярное моделирование ( реферат , курсовая , диплом , контрольная )
Таким образом, из всего выше сказанного, можно сделать вывод, что с помощью молекулярного моделирования стало возможным изучение десятков, а, возможно, в ближайшее время и сотен тысяч параметров каждой клетки. Выявляются тонкие различия экспрессии генов, а главное, выясняется, какие из генов, будучи экспрессированы в неправильное время, порождают изменение здоровой клетки до больной. Именно они имеют шанс стать теми мишенями, для которых фармацевтическая промышленность будет разработать лекарства, которые по-настоящему помогут от того или иного заболевания.
До сих пор большая часть лекарств была найдена, как ни странно, методом проб и ошибок, даже если речь шла об индустрии. Методом так называемого высокопроизводительного скрининга испытывались десятки, сотни, иногда под миллион веществ на определенных клеточных моделях тех или иных заболеваний, из которых десятки и сотни продвигались дальше как потенциальные лекарственные средства. К сожалению, в прошедшие годы крупные компании потратили миллионы долларов, проводя скрининг сотен тысяч веществ против определенных патологий, где ни одно из этих веществ не дало достойного кандидата. Компьютерный метод молекулярного моделирования позволяет одновременно исследовать десятки миллионов молекул на предмет связывания их с конкретной мишенью. Так как эти молекулы создаются против конкретной мишени, которой не должно быть у здорового человека, оказывается меньше шансов, что такая молекула станет токсичной и не пройдет дальнейшие этапы доклинических и клинических исследований.
Таким образом, создание таких технологий не просто ускоряет или удешевляет исследования, а зачастую позволяет в принципе найти хоть что-нибудь от важнейших заболеваний. Таким путем, например, совсем недавно были получены молекулы от вирусного гепатита. Впервые в истории человечества вирусный гепатит С стал если не излечимым, то контролируемым заболеванием. Для определенного вида рака груди появились очень эффективные, т.н. таргетные препараты, которые значительно повысили прогноз выживаемости больных. Компьютеры для разработки лекарств стали применяться лет 15 назад, но примерно последние 5 лет стали видны, наконец примеры, когда это стало по-настоящему получаться.
Молекулярное моделирование (ММ) — это собирательное название, относящееся к теоретическим подходам и вычислительным методам моделирования или изображения поведения молекул. Эти методы используются компьютерной химии, вычислительной биологии и науке о материалах для изучения молекулярных систем различных размеров. Простейшие вычисления могут быть выполнены вручную, но компьютеры становятся абсолютно необходимы при расчётах систем любого разумного масштаба. Общей чертой методов ММ является атомистический уровень описания молекулярных систем — наименьшими частицами являются атомы или небольшие группы атомов. В этом состоит отличие ММ от квантовой химии, где в явном виде учитываются и электроны. Таким образом, преимуществом ММ является меньшая сложность в описании систем, позволяющая рассмотрение большего числа частиц при расчётах. Молекулы могут быть смоделированы как в вакууме, так и в присутствии растворителя, например воды. Расчёты систем в вакууме называются расчётами "в газовой фазе", в то время как расчёты, включающие молекулы растворителя, называются расчётами "с явно заданным растворителем". Другая группа расчётов учитывает наличие растворителя оценочно, с помощью дополнительных членов в потенциальной функции — так называемые расчёты "с неявным растворителем". В настоящее время методы молекулярного моделирования стали обыденными при изучении структуры, динамики и термодинамики неорганических, биологических и полимерных систем. Среди биологических явлений, которые исследуются методами ММ, сворачивание белков, ферментативный катализ, стабильность белков, конформационные превращения и процессы молекулярного узнавания в белках, ДНК и мембранах.
3.3 Виртуальный скрининг
Виртуальный скрининг — это вычислительная процедура, которая включает автоматизированный просмотр базы данных химических соединений и отбор тех из них, для которых прогнозируется наличие желаемых свойств. Чаще всего виртуальный скрининг применяется при разработке новых лекарственных препаратов для поиска химических соединений, обладающих нужным видом биологической активности. В последнем случае процедура виртуального скрининга может быть основана либо на знании пространственного строения биологической мишени либо на знании структуры лигандов к молекуле данной биологической мишени. Ключевой процедурой виртуального скрининга, основанного на знании пространственной структуры биологической мишени, является молекулярный докинг, позволяющий предсказать пространственное строение комплекса "лиганд-белок" и исходя из него при помощи оценочных функций рассчитать константу связывания лиганда с белком. В этом случае из соединений, для которых предсказаны наибольшие значения констант связывания с молекулой белка, формируют сфокусированную библиотеку, из которой отбирают материал для дальнейшего биологического эксперимента. В качестве примера применения виртуального скрининга такого рода можно привести работу, направленную на поиск потенциальных лигандов NMDA- и AMPA-рецепторов
3.4 Программы для компьютерного моделирования
DockingServer предлагает простой в использовании веб-интерфейс, который управляет всеми аспектами молекулярной стыковки с лиганда и белка настройки. Ее дружественный пользователю интерфейс позволяет осуществлять расчеты и результаты оценки, проведенной исследователями из всех областях биохимии, DockingServer также обеспечивает полный контроль на установление конкретных параметров лиганда и белка настройки и стыковки и расчеты для более продвинутых пользователей. Приложение может использоваться для стыковки и анализа одного лиганда, а также высокую пропускную способность стыковка лиганда с белком-мишенью. DockingServer интегрирует большое количество вычислительных химических программ, конкретно направленных на правильность расчета параметров, необходимых на разных этапах стыковки процедуры, т.е. точной оптимизации геометрии лиганда, минимизации энергии, расчет заряда, расчет и стыковки белок-лиганд, комплексное представление. Таким образом, использование DockingServer позволяет пользователю осуществлять высокоэффективные и надежные стыковки расчетов путем интеграции ряда популярных программ, объединенных в одну веб-службу.
Программа Docking Server состоит из трех модулей содержащих следующие основные этапы стыковки расчетов
ü Лиганды можно напрямую загрузить из базы данных PubChem, либо в формате SDP файла
ü Пользователь может выбрать желаемый рН, влияющих на протонирование состояния лиганда.
ü Рассчитаная информация представляется пользователю в максимально удобном формате – в виде таблиц, списков. Для более опытных пользователей она может быть представлена в виде файла для самостоятельной работы
QuteMol — программа с открытым исходным кодом, для интерактивной визуализации молекулярных систем. QuteMol использует имеющиеся возможности современной компьютерной графики, используя библиотеку OpenGL. В программе доступен широкий набор графических эффектов. Методы визуализации QuteMol, направленные на улучшение реалистичности и облегчение восприятия 3D формы и структуры больших молекул или сложных белков.
Онлайн проект позволяющий каждому внести свой вклад в моделирование молекул лекарств от серьезных заболеваний. Все что нужно пользователю – это загрузить диструбтив программы и в свободное время оставить ее включенной на компьютере. Цель нашего текущего исследования состоит в том, чтобы разработать улучшенную модель внутри- и межмолекулярных взаимодействий и использовать эту модель для предсказания и проектирования макромолекулярных структур и взаимодействий. Приложения для предсказания и проектирования, которые могут представлять большой биологический интерес в их собственном праве, также обеспечивают строгие и объективные тесты, которые улучшают модель и увеличивают фундаментальное понимание. Мы используем компьютерную программу Розетта, чтобы выполнять вычисления белка и дизайна. В ядре Розетты имеются потенциальные функции для вычисления энергии взаимодействий в пределах и между макромолекулами, и методы для поиска структуры с самой низкой энергией для последовательности аминокислот (предсказание структуры белка) или комплекса белок-белок, и для поиска последовательности аминокислот с самой низкой энергией для белка или комплекса белок-белок (проектирование белка). Обратная связь от тестов предсказания и проектирования используется непрерывно, чтобы улучшить потенциальные функции и алгоритмы поиска. Развитие одной компьютерной программы для обработки этих разнообразных проблем имеет значительные преимущества: во-первых, различные приложения обеспечивают дополнительные тесты основной физической модели (фундаментальная физика / физическая химия, конечно, одна и та же во всех случаях); во-вторых, многие проблемы, представляющие текущий интерес, типа проектирования гибкого базового белка и стыковки белок-белок с базовой гибкостью, вовлекают комбинацию различных методов оптимизации.
Раздел: Медицина, здоровье
Количество знаков с пробелами: 47679
Количество таблиц: 0
Количество изображений: 2

Антон Чугунов, ПостНаука
Молекулярное моделирование – это способ моделировать на компьютере структуру и функции молекул. В первую очередь меня интересуют биологические молекулы.
Для изучения природы биологам очень важен эксперимент, поскольку он позволяет непосредственно изучить законы природы. Физики изучают физические законы, а биологи закономерности, которые есть в жизни. Зоологов интересует, как устроены животные, ботаников – растения, экологов – популяции животных и растений на Земле. Молекулярные биологи изучают, как между собой взаимодействуют молекулы и что в результате получается внутри живой клетки. Здесь бывают разные подходы. Биохимия интересуется химическими реакциями, превращениями одних молекул в другие, каким образом работает метаболизм, ферменты, как из ДНК получаются белки и так далее.
(Под биохимией я понимаю широкий спектр биохимических наук: это и молекулярная биология, и вирусология, и так далее.) А биофизике интересны физические законы: как могут провзаимодействовать между собой две молекулы, по каким законам это происходит? И можем ли мы эти законы вывести для себя и использовать их для теоретического моделирования?
Еще древние греки предположили, что материя состоит из атомов. В принципе, наверное, для этого было достаточно задуматься о том, из чего все состоит. Но до XX века существование атомов и молекул было до конца не подтверждено. Это связано с работами Бора и других физиков, которые показали уже несомненно, что есть атомы, и что есть молекулы. Уже к середине XX века прогресс был огромен. Уже открыли структуру биологических молекул, структуру ДНК, структуру многих белков (гемоглобина, например). В это время уже были заложены азы молекулярного моделирования. Первыми молекулярными моделистами были химики, которые рисовали структуру молекулы на бумаге. Уже можно было рядом нарисовать и представить себе две молекулы, как электронная плотность перемещается или как электрофилы атакуют нуклеофилы.
Всем нам нужно поучиться у Полинга, но, поскольку гением надо родиться, для большей плодотворности в наше время моделированием молекул занимаются на компьютере. Берут молекулярные редакторы, строят в них молекулы и запускают расчет, который позволяет нам проследить за тем, как молекула себя ведет. Допустим, мы можем смотреть молекулярную динамику, как существует белок в растворе или как две молекулы распознают друг друга. Как, допустим, маленькая молекула сначала плавает где-то в растворе, а потом взаимодействует с рецептором, и что-то происходит. Это, конечно, все очень сложно, и физика, лежащая в основе этих явлений, тоже очень сложна.
Автор – кандидат физико-математических наук, научный сотрудник лаборатории моделирования биомолекулярных систем Института биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова РАН.
В нашем первом посте про трехмерное моделирование вирусов мы перечислили основные стадии процесса и рассказали о том, с чего мы начинаем и как собираем исходную информацию. В этой заметке мы расскажем о следующем этапе работы — о создании моделей отдельных молекул, из которых впоследствии будет собрана целая частица.

Компоненты вирусной частицы Гриппа A/H1N1
Вирусная частица — это молекулярный механизм, решающий две принципиальные задачи. Во-первых, частица должна обеспечить упаковку вирусного генома и его защиту от деструктивных факторов среды, пока вирус путешествует из клетки, в которой он собрался, к клетке, которую он сможет заразить. Во-вторых, частица должна быть способна присоединиться к заражаемой клетке, после чего доставить вирусный геном и сопутствующие молекулы внутрь, чтобы запустить новый цикл размножения. Задач не очень много, поэтому вирусы, за редким исключением, могут позволить себе быть довольно экономными в том, что касается структуры.
В частности, геном большинства вирусов невелик и кодирует не очень много белков, нередко это число меньше 10. При этом вирус может заставить клетку синтезировать большое количество однотипных белков, из которых потом соберется вирусная оболочка — капсид. Таким образом, вирусные частицы обычно состоят из большого числа одинаковых элементов, которые связываются друг с другом как детали конструктора, часто образуя регулярные и симметричные структуры. Так, очень многие, хоть и не все вирусные упаковки или их фрагменты имеют спиральную или икосаэдрическую форму.

Примеры вирусных капсидов с икосаэдрической симметрией. Молекула бактриородопсина в правом нижнем углу — для сравнения. (Иллюстрация из обзора).
Для сборки модели вируса принципиально важно знать, как устроены отдельные белки общей структуры и как они друг с другом связываются, эту структуру формируя. Современная наука владеет целым набором методов, которые могут дать ответы на эти вопросы, однако ни один из подходов, к сожалению, не является универсальным и решает только часть задач которые стоят перед нами при создании научно достоверных моделей вирусов с атомной детализацией.
Белки: как получают, хранят и отображают информацию об их структуре?
Напомним, что белки — это полимерные молекулы, состоящие из последоватльно связанных между собой мономеров — аминокислот. В водных растворах белки обычно сворачиваются в сложные трехмерные глобулы (почти как головоломка “Змейка Рубика”), форма которых зависит от аминокислотного состава и некоторых других факторов. Пространственное строение этих глобул определяют в основном методами рентгеноструктурного анализа и ЯМР-спектроскопии. Также в последнее время к этой задаче позволяет подойти электронная микроскопия.
В целом, методы определения пространственной структуры молекул сложны и имеют целый набор ограничений, поэтому далеко не все вирусные белки описаны полностью. Так, рентгеноструктурный анализ предполагает наличие кристалла, через который пропускается рентгеновское излучение. Атомы кристалла провоцируют дифракцию рентгеновских лучей, по картине которой можно оценить распределение электронных плотностей в кристалле, а по этим данным уже восстановить расположения конкретных атомов. Этот метод дает разрешение вплоть до чуть более 1 ангстрема (0,1 нм), однако в случае белков проблема заключается в том, что далеко не все из них можно кристаллизовать. Особенно сложным это оказывается, если белок имеет гибкие подвижные или заякоренные в мембране фрагменты.
ЯМР-спектроскопия основана на явлении ядерного магнитного резонанса и позволяет описывать строение белков в растворе. Этот подход выявляет набор возможных положений атомов в молекуле и, в отличие от предыдущего метода, дает возможность оценить степень гибкости тех или иных ее участков. Но ЯМР-спектроскопия хорошо работает только для сравнительно небольших молекул, поскольку крупные белки дают слишком много шума.
Электронная микроскопия позволяет описать строение крупных молекулярных комплексов, что бывает очень полезно, когда речь идет о вирусах. Для многих симметричных структур можно получить большой набор изображений под разными углами, проанализировав которые можно воссоздать трехмерную картину. Для отдельных объектов разрешение, получаемое в результате применения разных вариантов электронной микроскопии (до 4-5 ангстрем), оказывается не многим хуже разрешения рентгеноструктурного анализа, хотя обычно для получения полной информации приходится совмещать разные подходы и, например, “вписывать” структуры отдельных белков в карты электронных плотностей, получаемые при помощи электронной микроскопии.

Структуры тримера белка оболочки ВИЧ (красные и голубые фрагменты молекул) в комплексе с участком одного из антител к этому белку (зеленые и желтые фрагменты), вписанные в карту электронной плотности, полученную методом крио-электронной микроскопии с разрешением 9 ангстрем. Из статьи Structural Mechanism of Trimeric HIV-1 Envelope Glycoprotein Activation.
Как мы писали в прошлом посте, получаемые структуры систематизируются и хранятся в базе данных Protein Data Bank. При этом в формате *.pdb записываются координаты атомов, и существует целый набор программ, позволяющих эти данные визуализировать и работать с такими структурами. Среди них, например VMD, Chimera, PyMol и десятки других.
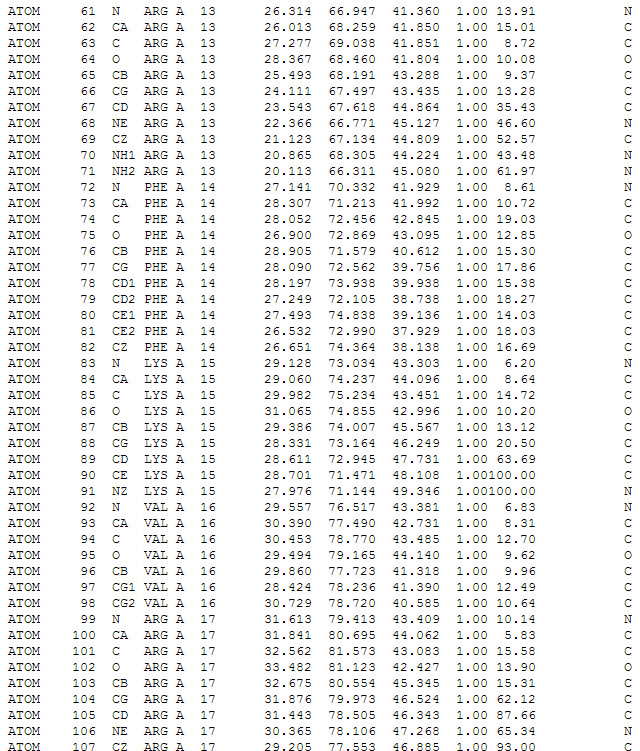
Скриншот текстового отображания файла в формате *.pdb. Описываются координаты отдельных атомов в аминокислотах белка.
Программы могут отображать белки несколькими способами. Помимо простого отображения атомов сферами разного диаметра, соответствующего ван-дер-ваальсовым радиусам атомов, существует возможность показать отдельные связи, поверхность молекулы, а также изгибы аминокислотной цепочки при помощи структур, напоминающих ленты (ribbon diagram), которые наглядно демонстрируют, где в белке аминокислоты образуют альфа-спирали, где бета-слои, а где неструктурированные участки.

Различные варианты визуализации структуры наружней части гемагглютинина вируса гриппа в программе Chimera.
В качестве отступления, надо сказать, что программы, в которых обычно работают ученые, визуализируя отдельные молекулы или белковые комплексы, чаще всего позволяют получить лишь довольно примитивные с эстетической точки зрения результаты (достаточно, например, посмотреть на несколько скриншотов из программы VMD). Принципиально более широкие возможности открываются, если импортировать модели молекул в программы, которые используют профессиональные дизайнеры и специалисты компьютерной трехмерной графики. Эти программы в сочетании с плагинами, улучшающими качество рендера, позволяют получать действительно интересные и привлекательные визуализации. Мы еще расскажем об этом в следующих постах. Пока просто приведем пример:
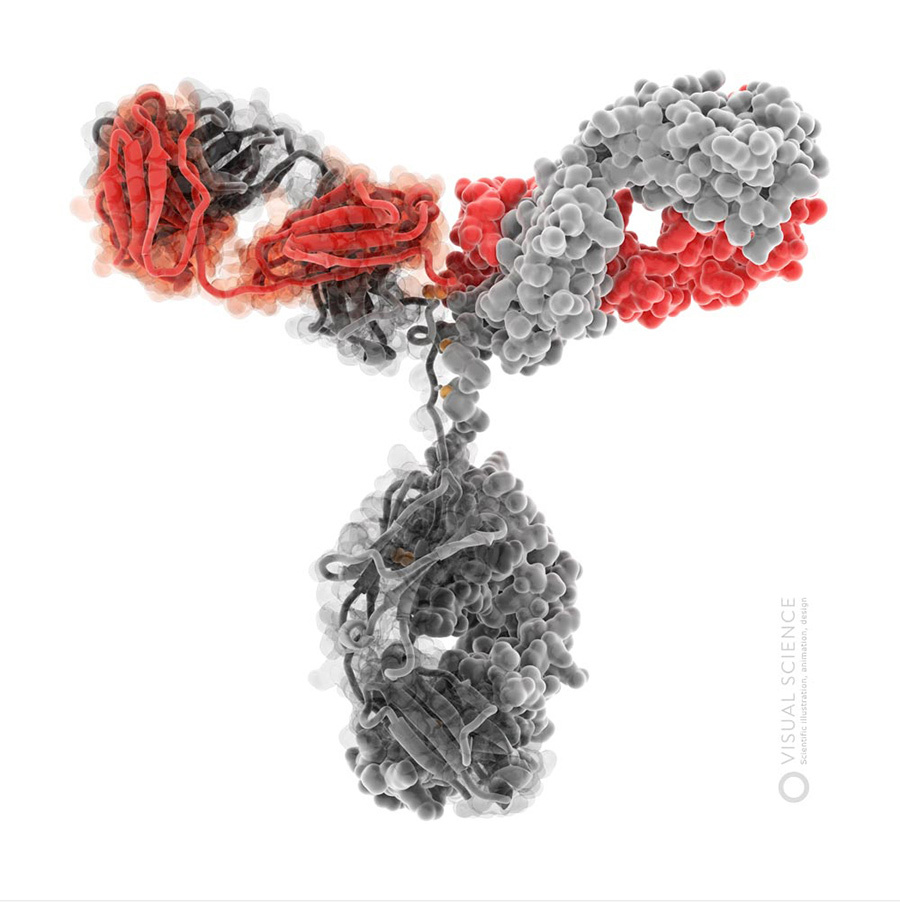

Изображения молекулы иммуноглобулина G.
Молекулярное моделирование

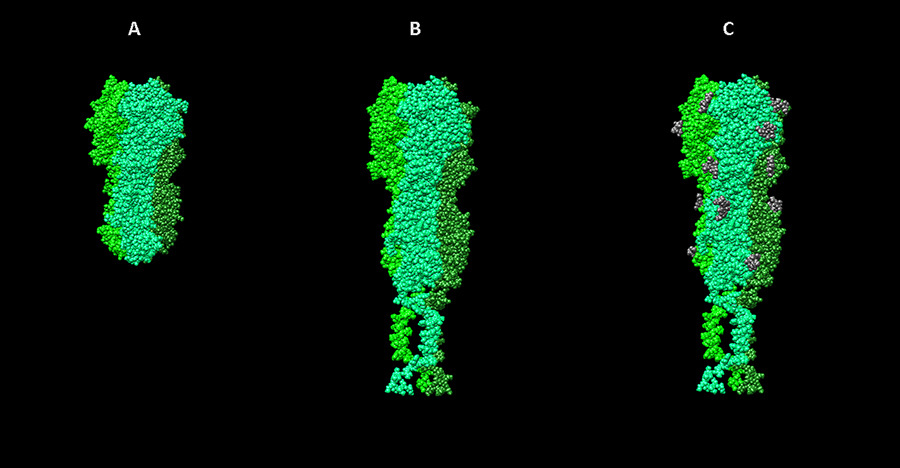
Шаблоны для моделирования нейраминидазного комплекса вируса гриппа. А — фрагмент мономера нейраминидазы N2 из структуры 2AEP в базе данных PDB, B — “стебель” гемагглютинин-нейраминидазы парагриппа (3TSI), С — трансмембранный пептид 2LAT. D — финальная полученная модель.
Окончательная модель белка обычно создается с учетом известных структур его фрагментов, найденных разными методами шаблонов, а также моделей от сервера I-Tasser. Для этого используется программа Modeller. Она позволяет строить модель по гомологии с использованием одного или нескольких шаблонов, а также вносить дополнительные модификации, например, создавать дисульфидные связи в заданных местах.
Докинг
Другим важным аспектом строения вирусов, информация о котором в научной литературе часто оказывается не полна, является взаимодействие между отдельными белками. В нашем случае от этого зависит то, какими поверхностями модели отдельных белков будут контактировать друг с другом и другими компонентами вириона в финальной модели. Информацию о взаимодействиях тоже позволяет уточнить структурная биоинформатика.
Программа докинга не моделирует естественный процесс образования комплекса, это было бы слишком медленно и ресурсоемко, а перебирает варианты взаимного положения двух или более молекул в поисках наилучшей структуры. При докинге обычно большую молекулу в комплексе называют рецептором, а меньшую — лигандом. Для определения качества структуры комплекса лиганда с рецептором используются различные оценочные функции. В идеале в качестве такой функции должна выступать свободная энергия системы, но она слишком сложно вычисляется, поэтому применяют различные эмпирические псевдопотенциалы, учитывающие потенциальную энергию (которая как раз вычисляется просто), площадь контакта лиганда и рецептора, соответствие различным правилам, которые исследователи вывели из анализа большого числа комплексов, и всякие загадочные слагаемые, не имеющие физического смысла, но улучшающие результат программы при испытании на большом количестве известных комплексов. Поиск минимума такого псевдопотенциала в современных программах обычно происходит с помощью различных вариаций метода Монте-Карло и генетических алгоритмов. В настоящее время существует множество программ молекулярного докинга (наиболее известные из них — Dock, Autodock, GOLD, Flexx, Glide), отличающиеся оценочными функциями, методами минимизации и дополнительными возможностями. При этом во время поиска молекулы рецептора и лиганда могут как оставаться неподвижными (такой тип докинга называется жестким), так и несколько менять конформацию (гибкий докинг). Очевидно, что второй вариант более ресурсоемкий, но и результаты такого поиска обычно правдоподобнее. Докинг малых молекул к белкам сейчас является стандартным этапом разработки новых лекарственных препаратов. Можно, например, провести докинги для 10 миллионов лигандов, и выбрать сотню наиболее перспективных соединений для дальнейшей экспериментальной работы — это называется виртуальный скрининг.
Помимо исследований небольших молекул, докинг может быть использован и для построения белок-белковых и белок-нуклеотидных комплексов. Для этих целей также разработано большое количество программ и онлайн-сервисов (ZDOCK, pyDOCK, HEX). Например, в ходе нашей работы над вирусом папилломы человека (ВПЧ) мы столкнулись с тем, что, несмотря на наличие полной структуры внешнего слоя капсида, образованнного белком L1, совершенно не было информации о строении белка L2, который в капсиде расположен ближе к геному, а соответственно, нет данных о том, как пентамеры L1 взаимодействуют с молекулами L2. Мы построили модель белка L2 по гомологии, используя сервер Tasser, после чего провели докинг в программе HeX. В ходе докинга роль рецептора выполнял пентамер L1. Именно на его поверхности проводился поиск оптимального места посадки L2. При этом все структуры оставались неподвижными. Т.е. использовался метод жесткого докинга. В результате была получена правдоподобная структура комплекса пентамера, собранного из L1 и минорного белка L2.
Посттрансляционные модификации
Наконец, биоинформатическими методами можно пытаться восстановить то, какие изменения в структуру вирусных белков вносит сама клетка, в которой они образуются. Большинство белков после синтеза подвергаются дополнительным химическим посттрансляционным модификациям (ПТМ), которые могут серьезно влиять на выполняемые белком функции. Среди таких модификаций фосфорилирование, убиквитинирование, гликозилирование, нитрозилирование, внесние разрывов и другие химические изменения. Многие поверхностные белки вирусов гликозилированы, причем эта модификация имеет непосредственное значение для выполнения основной функции поверхностных белков вируса — связывания с клеточными рецепторами. С другой стороны, белки вирусных матриксов — слоев, которые встречаются непосредственно под липидными оболочками некоторых вирусов, для заякоривания в мембране часто должны быть связаны, например, с миристиловой кислотой — небольшой гидрофобной молекулой, облегчающей взаимодействие белков с липидами. Таким образом, в нашей работе модификации белков тоже требуют внимания.
В настоящее время возможные ПТМ достаточно сложно предсказываются. Основные существующие методы и сервисы основаны на поиске соответствующей экспериментальной информации для сходных белков или поиске в последовательности исследуемого белка небольших участков, характерных для того или иного типа модификации.
В нашей работе при подготовке моделей мы пользуемся экспериментальной информацией, отраженной в соответствующей записи базы данных UNIPROT.
Стадии работы над моделью гемагглютинина вируса гриппа. А — визуализация структуры 3ZTJ из базы данных PDB. B — модель гемагглютинина вируса гриппа H1N1, построенная на основе гомологии с 3ZTJ с достраиванием трансмембранных участков молекулы. С — модель с учетом посттрансляционных модификаций (гликозилирования).
Молекулярная динамика и оптимизация структур
Последнее, о чем хочется упомянуть, — это то, что при подготовке новых моделей белков и, особенно, их комплексов, необходимо проводить оптимизацию структур. Наиболее простым методом оптимизации является минимизация энергии. Она используется для достаточно быстрого “спуска” системы в локальный минимум потенциальной энергии. Эту манипуляцию желательно проводить после каждой модификации структуры молекул. Она позволяет избежать таких неприятностей, как перекрывание атомов или появление неправильных длин связей. Различные методы минимизации энергии предусмотрены практически в любом программном пакете молекулярного моделирования.
Стоит отметить, что данный метод позволяет провести лишь предварительную и очень грубую оптимизацию. Для более точной подготовки пространственных структур используются методы молекулярной динамики или квантовой механики. Последние, например, используются для наилучшей оптимизации структуры небольших молекул лигандов и наиболее точных расчетов энергии межмолекулярных взаимодействий. Но, наибольшая точность, что вполне логично, связана с более ресурсоемкими вычислениями, что делает эти методы практически неподъемными в применении к большим биологическим макромолекулам.
Оценить поведение и стабильность структур достаточно массивных молекул, таких как полипептиды и нуклеиновые кислоты позволяют методы молекулярной динамики.
Метод молекулярной динамики заключается в изучении поведения атомов и молекул и их движений во времени. Расчеты молекулярной динамики позволяют, например, исследовать стабильность как отдельных молекул, так и их комплексов, позволяют оценить значимость возможных конформационных перестроек, влияние точечных мутаций и многое другое. Современные методы анализа результатов симуляций молекулярной динамики позволяют получить самые подробные сведения о поведении во времени как отдельных атомов, так и всей исследуемой системы.
В зависимости от того, насколько хорошо изучены белки того вируса, модель которого мы хотим создать, каждый раз приходится подбирать подходы для достройки и оптимизации моделей всех белков и их взаимодействий. После того, как все структуры получены, можно приступать к сборке полной модели. О том, как это делается, мы расскажем в следующих постах серии о создании научно достверных моделей вирусов человека.
PS:
Ставшая лидером в опросе прошлого поста тема Медицинская анатомическая иллюстрация — история изучения тела человека в работах иллюстраторов 5 столетий будет следующей. С потрясающими гравюрами, восковым моделями прошлого века, пластификатами трупов, атласами выдающся исследователей, 3Д реконструкциями на основе послойных срезов замороженного смертника, интерактивными приложениями и работами современных медицинских иллюстраторов. Скоро.
Читайте также: