
Гемолитико уремический синдром реферат
Обновлено: 17.05.2024
Гемолитико-уремический синдром (ГУС) хорошо известен многим педиатрам и в типичном случае проявляется остро развившейся диареей (гемоколит наблюдается у 75% больных), на фоне которой остро возникает состояние, сопровождающееся.
Гемолитико-уремический синдром (ГУС) хорошо известен многим педиатрам и в типичном случае проявляется остро развившейся диареей (гемоколит наблюдается у 75% больных), на фоне которой остро возникает состояние, сопровождающееся:
- микроангиопатической (неиммунной) гемолитической анемией (тест Кумбса отрицательный);
- тромбоцитопенией;
- острой почечной недостаточностью (ОПН).
Восстановление почечной функции в периоде выздоровления отмечается лишь у 70% больных, и связано это, прежде всего, с глубиной повреждения почечной ткани вследствие разнообразия причинно-значимых, провоцирующих факторов. Так, плохой прогноз имеют атипичные семейные (наследственные) и спорадические случаи ГУС, не ассоциированные с диареей, 25% этих больных погибают в острой фазе заболевания, у 50% прогрессирует отек-набухание головного мозга.
Этиология
Типичный ГУС вызывается шигаподобным токсином Stx1 Shigella disenteria и шигаподобным токсином Stx2 Escherichia coli O157:Н7. Цитопатический эффект шигаподобного токсина обнаружен на вероклетках почек африканских зеленых мартышек. У серотипа E. coli O157 имеются уникальные биохимические свойства — отсутствие ферментации сорбитола. Однако некоторые другие серотипы эшерихий способны вызывать диарею, ассоциированную с ГУС, у детей — О26, О145, О121, О103, О111, О113 и др. Они продуцируют другие токсины, отличные от шигаподобных токсинов своими субъединицами, аминокислотными последовательностями и молекулярным весом.
Классификация тромботических микроангиопатий (представлена Европейской педиатрической группой, 2006 г.).
С учетом этиологии:
- с включением инфекции — шигаподобный токсин S. disenteria и вероцитотоксин Е. coli.
Нарушения обмена веществ:
- генетические нарушения комплементарного обмена.
- образование аутоантител, включая аFH-АТ;
- нарушения метаболизма кобаламина.
Не полностью установленная этиология:
- ВИЧ;
- опухоли;
- лекарства;
- беременность;
- системная красная волчанка и антифосфолипидный синдром.
Установлено, что в основе не ассоциированного с диареей ГУС (non-Stx-HUS) имеет место генетическое нарушение — низкий уровень третьего компонента комплемента в сыворотке и нарушение его регуляции. Выявлены генетические маркеры, которые приводят к атипичному ГУС у больных с наследственной предрасположенностью. В настоящее время обнаружено более 50 мутаций в гене фактора НF1, кодирующих систему активации комплемента. В развитых странах такие больные проходят генетическое тестирование, а также определение уровня аутоантител. Так как атипичный ГУС протекает неблагоприятно с формированием в 50% случаев хронической почечной недостаточности (ХПН) или необратимого повреждения головного мозга, генетическое тестирование важно для решения вопроса о возможности успешной трансплантации почек таким пациентам [2].
Заболеваемость и факторы передачи
В Африке, Азии при бактериологическом исследовании кала от больных ГУС чаще высеваются серотипы шигелл, выделяющие Stx1, после его воздействия у 38–60% детей развивается гемоколит. В США ежегодно регистрируется до 70 тыс. заболевших эшерихиозом и примерно 60 летальных исходов. В Аргентине, Уругвае эшерихиоз эндемичен. Заболеваемость диареей, ассоциированной с ГУС, составляет 10 на 100 тыс. детей в год. Частое возникновение эшерихиоза связывают с традиционным употреблением мясных продуктов из телятины: до 40% молодых животных длительно выделяют в стуле Stx2 E. coli O157:Н7.
В России не ведется анализ заболеваемости диареей, ассоциированной с ГУС, у детей. Публикации скудны, осуществляются в основном реаниматологами. Диарея, ассоциированная с ГУС, этиологически редко расшифровывается. Врачи не диагностируют признаков тяжелого бактериального токсикоза в начальном периоде заболевания. Происходит недооценка степени тяжести состояния больных, соответственно, запоздалая адекватная терапия и неблагоприятные исходы.
Патогенез
- Stx1 S. disenteria и Stx2 E. coli O157 образуются в эпителиальных клетках слизистой кишечника.
- Токсинемия. Stx найден in vitro в эритроцитах, тромбоцитах, моноцитах, но в большей мере в нейтрофилах, которые имеют к нему специфический рецептор globotriaosylceramide Gb3.
- Проникновение Stx в эндотелий клубочков, рецепторы которых имеют в 100 раз более высокую аффинность, чем рецепторы нейтрофилов, в этой связи в кровеносном русле нет такого повреждения эндотелия, как в почках.
- Эндотелий мелких сосудов более чувствителен к Stx, чем эндотелий крупных сосудов (его рецепторы экспрессируются в 50 раз сильнее к Gb).
- Stx блокирует синтез протеинов в клетках, разрушая эндотелиальные клетки, индуцирует эндотелиальный апоптоз и лейкоцитоззависимое воспаление.
- В ренальных микрососудах моноцитами вырабатывается много туморнекротизирующего фактора, все это создает биохимическую базу для преимущественной локализации микроангиопатических повреждений в почках.
Таким образом, в детском возрасте у большинства детей встречается типичный или постдиарейный ГУС, который вторичен по отношению к острым кишечным инфекциям (ОКИ), а центральную роль в патогенезе почечных поражений, гемолиза и тромбоцитопении играет повреждение эндотелиальных клеток. В основе повреждения почек при ГУС лежит гломерулярная тромботическая микроангиопатия — утолщение стенки сосудов с отеком эндотелия и накоплением белков и клеточного детрита в субэндотелиальном слое в результате воздействия одного или нескольких повреждающих факторов. Кроме этого гистопатологические варианты ГУС включают в себя ишемию клубочков, которая в сочетании с тромбозом в последующем может приводить к многоочаговому или диффузному некрозу клубочков (коркового вещества), окклюзии клубочков фибриновыми тромбами.
Нормальный эндотелий обеспечивает эукоагуляционную ситуацию. Это поддерживается продукцией антитромбина III, простациклина, оксида азота, эндотелийзависимого релаксирующего фактора и т. д. При повреждении эндотелия его поверхность приобретает прокоагулянтные свойства, что в свою очередь способствует локальной активации свертывающей системы крови с внутрисосудистой коагуляцией, отложению фибрина в стенках и просвете капилляров. Это приводит к сужению или облитерации просвета капилляров клубочков, снижению скорости клубочковой фильтрации и уменьшению перфузии почечных канальцев с их вторичной дисфункцией или некрозом [5]. При ОКИ, осложненных ГУС, наиболее часто страдают внутриклубочковые сосуды, поражение которых возникает на ранних стадиях заболевания.
Генез тромбоцитопении при ГУС связан с усилением внутрипочечной агрегации тромбоцитов, при этом повышается уровень 3-тромбоглобулина и тромбофактора-4 — специфических тромбоцитарных белков, количество которых в плазме нарастает при активации тромбоцитов и снижении гломерулярной фильтрации [6]. Тромбоцитопении также способствует повышенное их потребление в тромбы. Кроме того, экспериментально показано, что после проведенной двусторонней нефрэктомии уровень тромбоцитов достаточно быстро восстанавливается. Это подтверждает причастность почек к данному лабораторному симптому.
Другим удивительным фактом является значимое снижение продукции эндотелиальными клетками простациклина (PGJ2) у некоторых больных с ГУС и членов их семей. Это предполагает наличие генетического дефекта, который может привести к развитию семейных случаев ГУС, при условии воздействия этиологического фактора на эндотелий сосудов.
Таким образом, при ГУС, обусловленным шигаподобным токсином, изменения наблюдаются непосредственно в клубочках и канальцах почек. Однако нефробиопсия, проведенная через несколько месяцев после заболевания, показывает, что большая часть клубочков сохраняет нормальное строение и только 15–20% склерозированы. Поэтому исходы ОКИ, осложненных ГУС, как правило, благоприятны, если своевременно купируется ОПН.
Основные клинические признаки (ОКИ + ГУС):
- острое начало, симптомы гастроэнтерита или тяжело протекающий колит, часто гемоколит (75% случаев);
- резкая бледность кожного покрова;
- кожный геморрагический синдром (петехии или пурпура);
- дизурия в виде олиго- либо анурии как основное проявление ОПН. При этом восстановление почечной функции возникает у большинства детей (70% случаев), а у 30% больных наступает либо смерть в результате развития синдрома полиорганной недостаточности, либо формирование ХПН.
Дополнительные симптомы ОКИ + ГУС:
- анорексия;
- раздражительность;
- гипертензия;
- спленомегалия;
- желтуха, темный цвет мочи (гемоглобинурия);
- признаки застоя в системе кровообращения (отек легких, кардио-, гепатомегалия, расширение вен, тахикардия).
Лечение больных с ГУС проводится исключительно симптоматическое, поддерживающее, поскольку патогенетической терапии с доказанной эффективностью в настоящее время не существует.
Ниже представлены рекомендации по ведению пациентов с ГУС, опубликованные Европейской педиатрической исследовательской группой [3] и в Консенсусе исследовательской группы по печеночно-почечной трансплантации при ГУС.
Лечение:
- высококачественная диета;
- при выраженной анемии переливание эритроцитарной массы;
- инфузии плазмы, включая плазмообмен;
- перитонеальный диализ;
- гемодиализ при стойкой тяжелой ОПН;
- в терминальной стадии хронический диализ с перспективой трансплантации почки.
Нами проводилось изучение клинического профиля, спектра функциональных нарушений, прогностических факторов и исходов у 25 детей с ОКИ, осложненными гемолитико-уремическим синдромом, которые находились на лечении в МУЗ ДГКБ № 3 Новосибирска в период с 1991 по 2010 гг.
Наибольшее количество случаев (16 из 25 больных) ГУС наблюдалось у детей в возрасте до трех лет, что согласуется с данными литературы [1, 3]. В 1,3 раза чаще заболевание развивалось у девочек, такое соотношение встречается не везде, например, в Непале мальчики болеют в 3 раза чаще, чем девочки [4].
В первые трое суток от начала ОКИ ГУС развился у 13 больных, что составило 52% больных, до 5 дней — у 7 (28%) больных, и от 6 до 8 дней — у 5 (20%) детей. Таким образом, острое начало ГУС отмечалось только у половины, а у остальных больных проходило некоторое время от начала диареи, болезнь манифестировала клиникой гастроэнтероколита, поэтому терапия продолжалась на участке и была неадекватной степени тяжести. При этом длительно использовались сорбенты, не назначались антибактериальные препараты, либо использовался фуразолидон без эффекта, и терапия не менялась до появления клинических симптомов ГУС.
Результаты бактериологических исследований кала были положительны лишь у 8 больных. Так, в периоде диареи у двух пациентов в копрокультуре отмечался высев шигелл Флекснера; у одного — Salmonella typhy murium; у двух — E. coli O26; у трех — E. coli O157. Сложность верификации E. coli O157, выделяющей Stx2, обусловлена уникальным свойством ферментации бактерий данного серотипа на средах, содержащих сорбитол.
Развернутая клиническая картина в начале заболевания включала лихорадку, рвоту, абдоминальные боли, одышку, при этом колит отмечался у всех 100% заболевших, а гемоколит — только у пяти больных. Мочевой синдром в виде макрогематурии в острой фазе болезни имел место у двух детей.
Поражение центральной нервной системы (ЦНС) диагностировано у 14 детей (оглушение, сопор, судороги, кома), что было расценено нами как проявление токсикоза, гипергидратации, метаболических изменений (ацидоза), электролитных нарушений — гиперкальциемии, гипокалиемии (гиперкалиемия выявлялась редко), гипонатриемии, нарушение осмолярности плазмы. Гипонатриемия была связана как с потерей этого электролита с кишечником, так и почками. На фоне олигурии экскреция натрия усиливается за счет торможения реабсорбции в канальцах. Начальные признаки поражения ЦНС — повышенная возбудимость, беспокойство, затем прогрессирующая вялость, затем больные впадали в кому. Нарушение сознания проявлялось в основном у детей первых лет жизни.
Основные показатели системы гемостаза, за исключением тромбоцитопении, изменялись незначительно. Это свидетельствовало о том, что в патогенезе ГУС отсутствует системное тромбообразование (ДВС-синдром (диссеминированное внутрисосудистое свертывание)), а тромбоз сосудов осуществляется преимущественно на уровне почек. Возможно, у детей и есть период диссеминированного свертывания крови, но факторы, потребляемые в этот период заболевания, быстро восстанавливаются до нормы. Рекомендуется поддерживать нормальный уровень фибриногена, факторов свертывания введением свежезамороженной плазмы, где они содержатся в большом количестве. Так, у наших больных время свертывания, АЧТВ (активированное частичное тромбопластиновое время), уровень фибриногена, количество РФМК (растворимых фибрин-мономерных комплексов), как правило, соответствовало контрольным значениям.
Однако кровотечения отмечались у двух больных. В 1997 г. мы наблюдали случай ГУС у ребенка Р. 3 лет с шигеллезом Флекснера и язвенно-некротическим поражением толстого кишечника. В данном случае отмечался тяжелый гемоколит с периодическими кишечными кровотечениями в течение 12 дней. Предпринимались попытки коррекции гемостаза путем введения большого количества свежезамороженной плазмы, но у больного развился парез кишечника, который определил наступление летального исхода. В 2003 г. у больной К. 6 лет наблюдалось массивное кровотечение из толстого кишечника, использование препарата NovoSeven — рекомбинантного фактора свертывания крови VIIa позволило остановить кровотечение, в последующем наступило выздоровление. Тромбомасса вводилась некоторым больным с критическими цифрами, но только при кровотечениях, поскольку срок жизни тромбоцитов ограничен.
Значения сывороточного уровня мочевины и креатинина в остром периоде болезни значительно превышали контрольные возрастные показатели (в среднем в 1,5–2 раза и более). Тяжесть ГУС обусловливает глубина поражения почек, соответственно, состояние больного и исход зависят от снижения почечных функций. Как известно, длительность анурии предопределяет прогноз — чем дольше она продолжается, тем вероятность восстановления почечной функции меньше и чаще наблюдается переход в ХПН [3]. При сохранении водовыделительной функции почек (так называемая неолигурическая ОПН), даже при высоких цифрах мочевины, прогноз, как правило, благоприятен. Для олигоанурической стадии ГУС не характерны отеки в связи с потерями жидкости со стулом и перспирацией. Протеинурия встречалась у 38% и микрогематурия — у 70% больных.
Варианты заместительной почечной терапии
Плазмообмен — первый уровень терапии, он выполнялся всем 25 больным в объеме 1,5–2 объема циркулирующей плазмы. Это коррекция плазменных факторов, осуществлялась в остром периоде ежедневно, в дальнейшем по необходимости. Острый период ГУС у всех детей протекал по-разному — обычно в течение 2–5 дней наблюдались токсикоз, тромбоцитопенические сыпи, тромбозы катетеров. Если за это время не восстанавливался диурез, больному устанавливали перитонеальный катетер и проводили перитонеальный диализ от 2 до 6 недель.
Перитонеальный диализ выполнялся шести больным (четырем в сочетании с гемодиализом и двум — с гемодиафильтрацией). Есть больные без тяжелого токсикоза и выраженных электролитных нарушений, которым можно проводить только перитонеальный диализ, и они поправляются. Перитонеальный диализ — щадящая заместительная терапия, при которой медленно происходит обмен жидкости через брюшину, он может использоваться длительно, до восстановления функции почек.
С 1991 по 2003 г. плазмообмен в сочетании с гемодиализом был выполнен 14 (56%) больным. С 2008 г. для стабилизации гомеостаза в стационаре осуществляется гемодиафильтрация в сочетании с перитонеальным диализом (использовалась в лечении трем больным). Принцип гемодиафильтрации — замена интерстициальной жидкости на специальные растворы, которые полностью, кроме белков, соответствуют составу плазмы. При гемодиафильтрации плазмообмен осуществляется аппаратным способом, это многочасовая процедура, иногда она длится сутки, при этом диализирующая жидкость вводится со скоростью 70 мл/мин. Этот метод терапии более эффективен при SIRS-синдроме. Гемодиализ показан больным с нарушением азото- и водовыделительных функций почек без симптомов воспаления, снижает уровень азотемии, нормализует гомеостаз за 3–4 часа, поток диализирующего раствора по фильтру составляет 500 мл/мин.
В нашем наблюдении олиго/анурия у трех больных отсутствовала, а в остром периоде болезни доминировали симптомы гемолиза, у трех пациентов олиго/анурия сохранялась до трех суток, у 8 детей — до 8 суток, у шести человек до 15 суток и у двух — до 20 и более. Так, у пациента с длительностью олигурической стадии ОПН 28 дней, которому проводилась почечная заместительная терапия, полностью восстановились функции почек. У другой больной, после 42 дней заместительной почечной терапии, развилась ХПН.
Таким образом, при современном и адекватном лечении ГУС у детей, который возник после перенесенной ОКИ, исходом в 18 (72%) случаях стало выздоровление, в одном случае (4%) переход в ХПН, в 6 (24%) — смерть больных. К прогностически неблагоприятным признакам можно отнести:
- длительную анурию;
- нарушения со стороны ЦНС;
- остро развивающийся массивный гемолиз эритроцитов;
- гиперлейкоцитоз;
- гиперкалиемию;
- гемоколит, неразрешающийся парез кишечника.
Литература
- Байко С. В. Гемолитико-уремический синдром: эпидемиология, классификация, клиника, диагностика, лечение (Обзор литературы. Часть 1) // Нефрология и диализ. 2007, т. 9, № 4, с. 370–377.
- Байко С. В. Гемолитико-уремический синдром: эпидемиология, классификация, клиника, диагностика, лечение (Обзор литературы. Часть 2) // Нефрология и диализ. 2007, т. 9, № 4, с. 377–386.
- Ariceta G., Besbas N., Johnson S., Karpman D. et al. Guideline for the investigation and initial therapy of diarrhea-negative haemolytic uremic syndrome // Pediatr. Nephrol. 2009, 24, s. 687–696.
- Cerda J., Bagga A., Kher V., Chakravarthi R. M. The contrasting characteristics of acute kidney injury in developed and developing countries // Nature Clinical Practice Nephrology. 2008, 4, s.138–153.
- Petruzziello T. N., Mawji I. A., Khan M., Marsden Ph. Verotoxin biology: molecular events in vascular endothelial injury // Kidney International. 2009, 75, s. 17–19.
- Tarr Ph. Shiga toxin-associated hemolytic uremic syndrome and thrombotic thrombocytopenic purpura: distinct mechanisms of pathogenesis //Kidney International. 2009, 75, s. 29–32.
Е. И. Краснова, доктор медицинских наук, профессор
С. А. Лоскутова, доктор медицинских наук, доцент
О. В. Гайнц

В 1955 г. C. Gasser с соавторами описали наблюдавшееся ими у 5 детей заболевание, которое представляло собой сочетание острой почечной недостаточности (ОПН) с гемолитической анемией и тромбоцитопенией, развивающимися на фоне инфекционной диареи (E. coli, Sh. dysenteriae, S. pneumoniae), и назвали его гемолитико-уремическим синдромом (ГУС).
Эпидемиология
С данным синдромом должен быть знаком каждый педиатр, ведь ГУС является основной причиной развития ОПН у детей до 3-х лет. Частота возникновения ГУС у детей данного возраста составляет 2–3 случая на 10 тысяч детей. Причем в разных регионах заболеваемость типичным ГУС (тГУС) значительно варьирует (в зависимости от численности сельского населения, особенностей водоснабжения — в Аргентине и Уругвае эшерихиоз эндемичен, поэтому частота достигает 10 случаев на 100 тыс. населения в год; в более холодных регионах заболеваемость также выше — в Шотландии, по сравнению с Англией, в 2 раза выше — 3,4 vs 1,54 на 100 тыс. населения в год). Для тГУС чаще характерны эпидемические подъемы заболеваемости, но могут быть и спорадические случаи (более характерно для атипичного ГУС). Резервуаром инфекции являются фекалии крупного рогатого скота (E. coli O157 длительно выделяют в стуле Shiga like toxin (Stx) 2 типа). Человек заражается при употреблении сырой телятины, непастеризованного молока, загрязненных фруктов и овощей, контаминированной воды из колодца и водоемов, а также при неисправностях водопровода. Прямой контакт детей с животными или их испражнениями и передача от человека к человеку являются не менее важными механизмами передачи этой инфекции.
Классификация
Официальной классификации ГУС нет. По причинам возникновения выделяют инфекционные и неинфекционные формы (рис. 1). К инфекционным формам ГУС относят:
- ГУС, ассоциированный с шига-токсином (Sh. dysenteriae тип 1);
- ГУС, ассоциированный с микроорганизмами, секретирующими нейраминидазу (S. pneumoniae);
- ГУС, ассоциированный с ВИЧ-инфекцией, и др.
К неинфекционным формам относят идиопатический ГУС, наследственный ГУС (связанный с аномалиями ADAMTS-13), лекарственно-индуцированный ГУС (прием ингибиторов mTOR или ингибиторов VEGF) и другие формы.

Рисунок 1 | Классификация ГУС
Помимо вышеописанной классификации, ГУС можно отнести к первичным тромботическим микроангиопатиям (ТМА), этиология и патогенез которых установлены:
- ГУС, индуцированный инфекцией или ассоциированный с диареей (тГУС, ГУС-(D+));
- Атипичный ГУС, обусловленный генетическими нарушениями или изменениями иммунной системы, приводящими к патологии системы комплемента (аГУС, ГУС-(D–));
- Тромботическая тромбоцитопеническая пурпура (ТТП, болезнь Мошковица), связанная с аномалиями фермента ADAMTS-13 (врожденная или приобретенная).
Патогенез
Типичный ГУС
Основным фактором, инициирующим развитие тГУС, который обусловливает до 80 % от общего числа случаев заболевания, является энтерогеморрагическая кишечная палочка (E. coli, EHEC, серотип О157:Н7), синтезирующая шигаподобный токсин (веротоксин 1 и/или 2 типа). Данный штамм обладает высокой патогенностью для человека (для заражения достаточно 10 3 микроорганизмов), однако диарея развивается только в каждом 10-м случае (рис. 2).

Рисунок 2 | Патогенез тГУС, ассоциированного с EHEC
После попадания E. coli в кишечник она связывается с ворсинками подвздошной кишки и эпителиальными клетками пейеровых бляшек при помощи специального белка, вызывая в конечном итоге гибель клеток с развитием диареи, переходящей в гемоколит (его возникновение связано с веротоксином, который способен повреждать сосуды слизистой оболочки кишечника). Шигаподобный токсин (SLT, Stx), высвобождающийся из кишечника, попадает в печень, где часть его метаболизируется, а другая часть попадает в системный кровоток, вызывая повреждение эндотелия органов-мишеней (легкие, почки, головной мозг).
SLT транспортируется в крови в основном нейтрофилами, но может перемещаться по системному кровотоку и при помощи моноцитов, тромбоцитов и/или их комплексов (липополисахарид кишечной палочки связывается с тромбоцитами, вызывая их активацию и агрегацию). За счет субъединицы В Stx имеет высокое сродство к мембраносвязанным гликосфинголипидам — Gb3/Gb4-рецепторам (в 100 раз выше, чем таковое с нейтрофилами).
Помимо этого, ЛПС, концентрация которого в крови прямо коррелирует с таковой у шига-токсина, обусловливает повышенную продукцию провоспалительных цитокинов — интерлейкинов 1, 6, 8, а также фактора некроза опухоли-альфа (TNF-α). Те, в свою очередь, повышают экспрессию рецепторов на мембранах связывающих их моноцитов, приводя тем самым к более выраженному токсическому эффекту Stx.
Патогенез ГУС, вызванного Shigella dysenteriae 1 типа, схож с таковым у E. coli (рис. 3, 4, 5). Однако этот тип ГУС протекает тяжелее, чем ГУС, ассоциированный с шигаподобным токсином E. coli. Связано это, скорее всего, с липополисахаридным эндотоксином шигелл, который путем сложного взаимодействия с рецепторами TLR4 (на мембранах клеток) и NLR 1, 2 (Nod like receptors, расположены внутриклеточно) вызывает активацию сигнального пути NF-kB, что, в свою очередь, приводит к массивному выделению интерлейкина 8, являющегося мощным хемокином для нейтрофилов, макрофагов и лимфоцитов. Активированные нейтрофилы путем массивного выброса воспалительных цитокинов, помимо повышения секреции специфических рецепторов на мембранах эндотелиоцитов, вызывают активацию перекисного окисления липидов (ПОЛ), приводящего к повреждению не только эндотелия, но и эритроцитов, а также активацию лизосомальных ферментов, например, эластазы или α1-антитрипсина, которые также усугубляют эндотелиальное повреждение.

Рисунок 3 | Патогенез тГУС, ассоциированного с Shigella dysenteriae
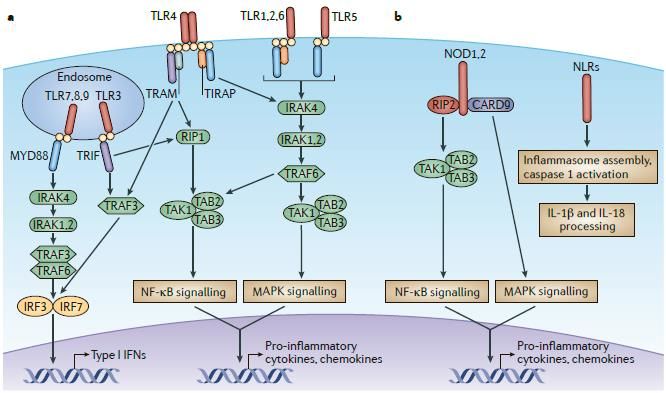
Рисунок 4 | Рецепторы TLR4 и NOD 1-2, через которые Shigella dysenteriae активирует сигнальный путь NF-kB
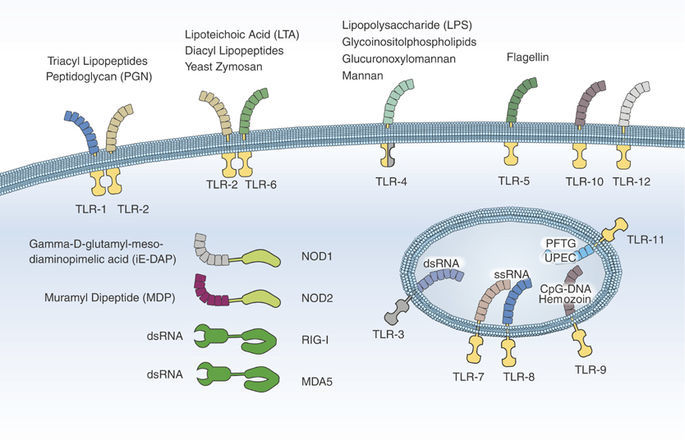
Рисунок 5 | Молекулярная основа патогенеза тГУС, ассоциированного с Shigella dysenteriae
Рисунок 6 | Патогенез тГУС, ассоциированного со Streptococcus pneumoniae
Атипичный ГУС
Патогенез атипичного ГУС (аГУС) разительно отличается от тГУС. В его основе лежат мутации регуляторных белков системы комплемента (чаще всего аГУС ассоциирован с мутацией комплементарного фактора Н (CFH), на втором месте располагается мембранный кофакторный протеин (MCP), тройку замыкает комплементарный фактор I (CFI)).
При активации комплемента образуется C3-конвертаза, расщепляющая C3 на малый (С3а) и большой (C3b) фрагменты, который и опсонизируется на поверхности микробной клетки и формирует мембраноатакующий комплекс (МАК), состоящий из C5b, C6, C7, C8 и C9, что приводит к осмотическому лизису этой клетки. Для того, чтобы активированная система комплемента не уничтожила собственные клетки, на их поверхности расположены белки-регуляторы (DAF и CR1); помимо этого, часть таких белков синтезируется в печени и циркулирует в плазме крови в неактивном состоянии. К таким белкам относят комплементарный фактор H (CFH), фактор I (CFI) и мембранный кофакторный протеин, закрепленный на поверхности клеток (CD46). Фактор I, главный из вышеперечисленных факторов, расщепляет C3b и C4b. Фактор Н и CD46 являются кофакторами фактора комплемента I (рис. 7). Первый из них связывается с гликозаминогликанами собственных клеток организма, отсутствующими на мембранах бактериальных клеток, а также ингибирует активность C3-конвертазы. При мутации данных регуляторных белков происходит утрата защиты эндотелиальных клеток от повреждения конечными продуктами активации альтернативного пути комплемента (рис. 8).

Рисунок 7 | Нормальная регуляция системы комплемента

Рисунок 8 | Патогенез аГУС
Клиническая картина
В течении тГУС условно можно выделить 2 периода. Продромальный период характеризуется диареей, в ⅔ случаях диарею может сопровождать рвота. Гемоколит, характерный для тГУС, ассоциированного с шига-токсином, обычно развивается у каждого третьего больного через 2–3 дня после появления диареи.
Ввиду частой гиподиагностики адекватный контроль регидратации может отсутствовать, поэтому дополнительным признаком тГУС у детей можно считать артериальную гипертензию, которая отличается упорным течением и плохо поддается терапии. После восстановления нормального диуреза может отмечаться второй подъем АД, связанный с избыточной выработкой ренина.
Поражение ЦНС в виде генерализованных судорог, нарушения сознания вплоть до комы развивается в каждом четвертом случае и характеризует тяжесть течения тГУС.
Клиника аГУС имеет ряд особенностей!
Для аГУС характерно очень раннее начало (начиная с 2 месяцев при мутации CFI, c 6 месяцев при мутации CFH). Чаще всего аГУС манифестирует неспецифическими симптомами в виде общей слабости, недомогания без предшествующей этим симптомам диареи (при ее наличии затрудняется дифференциальный диагноз с тГУС). Возможно развитие гриппоподобного синдрома. У взрослых клиника может развиваться стерто, при этом классическая триада ГУС имеет слабую выраженность. Возможно развитие неполной триады без тромбоцитопении. Также для аГУС характерен семейный характер заболевания (в 25 % случаев имеется отягощенный наследственный анамнез). Большинство пациентов имеют выраженный отечный синдром вплоть до анасарки даже при отсутствии нефротического синдрома и ОПН. Также для больных с аГУС характерна АГ, связанная с перегрузкой объемом при манифестирующей ОПН или возникающая вследствие активации РААС, вызванной ишемией почечной ткани, обусловленной тромботической микроангиопатией. В отличие от тГУС, для атипичной формы характерно развитие кардиомиопатии с признаками острой СН. Описаны случаи развития острого панкреатита вплоть до панкреонекроза. В общем и целом клиника аГУС отличается яркой вариабельностью симптомов, что затрудняет ее своевременную диагностику (характерно рецидивирование).
Диагностика
Лабораторная диагностика как типичного, так и атипичного ГУС основана на выявлении признаков тромботической микроангиопатии:
- гемолитическая анемия — уровень гемоглобина ниже 90 г/л, выраженный ретикулоцитоз. Помимо этого, в крови могут появляться остатки эритроцитов — шизоциты (больше 1 %). Разрушение эритроцитов происходит из-за механического повреждения нитями фибрина при их прохождении через тромбированные сосуды почечных клубочков;
- тромбоцитопения ниже 150 тыс/мм 3 ;
- выраженный лейкоцитоз выше 20 х 10 9 /л; характеризует тяжесть ГУС.
Помимо этого, к неспецифическим признакам гемолиза эритроцитов можно отнести повышенный уровень ЛДГ, уменьшение уровня свободного гаптоглобина, гипербилирубинемию (за счет непрямой фракции). При проведении прямой пробы Кумбса результат будет отрицательным как у тГУС, так и аГУС.
Система диагностики ОПН, согласно критериям AKIN, основана на исследовании концентрации креатинина в сыворотке крови, СКФ, рассчитанной по формуле MDRD или CKD-EPI, а также на объеме мочи в течение суток. Для достоверной диагностики анурической стадии ОПН при подозрении на ГУС возможно выявление ранних признаков почечного повреждения (повышение уровня цистатина С, NGAL), а также повышения уровня калия крови выше 6 ммоль/л.
С целью подтверждения ГУС, связанного с шига-токсином, при наличии симптомов со стороны ЖКТ необходимо провести:
- посев кала на среду МакКонки для выявления серотипа E. coli O157:H7;
- определение шига-токсина в кале методом ПЦР или его выявление в сыворотке крови;
- возможно определение антител к липополисахариду эндемичного для данного региона серотипа E. coli.
Для исключения тромботической тромбоцитопенической пурпуры (ТТП) всем больным с характерной для ГУС клинической картиной необходимо определение активности ADAMTS-13 (менее 5 %). Для пациентов с аГУС типично снижение данного показателя, однако он в любом случае будет выше 10 % (в норме составляет 80–110 %).
Если при госпитализации больного в его анамнезе были выявлены предшествующие тромботические микроангиопатии, необходимо исследовать кровь на содержание С3 и С4 компонентов комплемента, а также на аутоантитела к фактору Н (анти-FH-антитела). Помимо этого, необходимо проведение дифференциальной диагностики со системными заболеваниями соединительной ткани (рис. 9). Развитие характерного симптомокомплекса во время беременности требует исключения специфической акушерской патологии.

Рисунок 9 | Дифференциальная диагностика ГУС
Лечение
Относительно специфическое лечение разработано только для атипичной формы ГУС. В настоящее время единственным допущенным до применения ингибитором системы комплемента является экулизумаб (рекомбинантное моноклональное антитело против компонента комплемента С5). Данный препарат блокирует расщепление С5 компонента комплемента (С5а — провоспалительный, C5b — протромботический компонент) и формирование на мембране собственных клеток МАК C5b-9.
Клинические испытания показали, что в профилактике и лечении аГУС экулизумаб оказался более эффективен, чем плазмаферез. Однако наилучшей схемой лечения считается введение препарата на фоне проведения плазмафереза, так как последний удаляет часть препарата из циркуляции, тем самым предотвращая развитие побочных реакций.
Введение свежезамороженной плазмы (СЗП) не предотвращает развитие терминальных стадий ХБП у больных с аГУС. Ее использование оправдано ввиду того, что СЗП является источником нормальных комплементарных факторов CFH и CFI. Как и в ситуации с экулизумабом, введение СЗП лучше сочетать с плазмаферезом (удаляются мутантные комплементарные факторы и анти-CFH антитела; удаление части плазмы предотвращает гиперволемию и следующую за этим острую сердечную недостаточность). Ввиду генетической детерминированности аГУС высок риск развития рецидива. Это, в свою очередь, требует проведение адекватной профилактической терапии, включающей в себя санацию очагов хронической инфекции, а также проведение своевременной вакцинопрофилактики. Трансплантация почки как метод лечения аГУС не имеет на данный момент широкой доказательной базы (описано всего 3 случая пересадки). Риск возврата аГУС сразу после трансплантации чрезвычайно высок, особенно у пациентов с мутацией комплементарного фактора Н.
Консервативное лечение тГУС заключается в проведении корректной регидратационной терапии. Как говорилось выше, неспецифичность клинической картины обусловливает высокий процент гиподиагностики, в связи с чем следующая за этим попытка коррекции водно-электролитного баланса оказывает отрицательный эффект (например, перегрузка объемом приводит в итоге к развитию острой сердечной недостаточности и активации РААС (гиперренинемии) — формируется резистентная к проводимой терапии артериальная гипертензия. Использование петлевых диуретиков, например, фуросемида, не оправдано; предпочтение стоит отдавать гемодиализу (на фоне гиперкалиемии или метаболического ацидоза) ввиду того, что диализ чаще всего начинается в олигоанурической стадии ОПН. При развитии анемии тяжелой степени (Hb ниже 70 г/л) показано переливание эритроцитарной массы. Антибиотики не являются основным компонентом терапии тГУС. Однако раннее назначение цефалоспоринов III поколения или фторхинолонов снижает риск развития тГУС, ассоциированного с S. dysenteriae типа 1.
Гемолитико-уремический синдром (болезнь Гассера) является одним из немногих приобретенных заболеваний, встречающихся обычно в детской возрастной группе. Вероятно, данный синдром охватывает целый спектр заболеваний, варьируя от классической саморазрешающейся формы у детей грудного возраста до более грозной и часто фатальной тромбоцитопенической тромбо-гемолитической пурпуры (болезнь Мошкович), наблюдаемой у взрослых. Для этих заболеваний характерна общая триада: острая почечная недостаточность с олигурией, гемолитическая анемия и тромбоцитопения. Однако заболевания, протекающие под прикрытием болезни Гассера, могут также иметь ту или иную степень артериальной ангиопатии, гипертензии и поражения ЦНС, кишечника или печени.
Хотя общий знаменатель, который связывал бы различные формы болезни или служил базисом для их классификации или выбора лечения, еще не найден, некоторые из недавних основных научных достижений помогают проникнуть в суть данной проблемы.
Повреждение эндотелиальных клеток играет центральную роль в патогенезе почечных поражений, гемолиза и тромбоцитопении. Повреждение эндотелия может ограничиваться капиллярными мембранами, но не исключено и вовлечение артерии. Такое разграничение может помочь в более детальном классифицировании и прогнозировании, а также объяснении природы большой группы симптомов. Механизм повреждения эндотелия в настоящее время является объектом споров и научных исследований. Идентифицировано по меньшей мере 5 агентов, способных оказывать повреждающее действие на эндотелиальные клетки при болезни Гассера: эндотоксин, нейраминидаза, эстроген - содержащие контрацептивы, истинный цитотоксин (веротоксин), продуцируемый некоторыми штаммами кишечной палочки, и токсин, вырабатываемый бактериями Григорьева - Шиги. Во время эпидемий Гемолитико - уремического синдрома было выделено несколько видов вирусов, чаще всего Коксаки, ECHO и аденовирус, но без четкой связи с заболеванием. Более убедительным является развитие данного синдрома у больных с инфекциями, вызванными шигеллой, сальмонеллой, цитотоксической, кишечной палочкой или пневмококком. В настоящее время внимание исследователей сосредоточено на веротоксин - продуцирующей E. Coli (VTEC O157:H7) и Shigella disenteriae type1. Болезнь Гассера может также наблюдаться у женщин, принимающих гормональные контрацептивные средства, содержащие эстроген, или у беременных. Некоторые исследования отмечают высокую частоту возникновения Гемолитико - уремического синдрома при использовании ампициллина для лечения дизентерии вызваной Shigella disenteriae type1.[3]
Повреждение эндотелиальных клеток затрагивает и приводит в действие целый ряд вторичных процессов: локальное внутрисосудистое свертывание крови, слипание тромбоцитов и агрегацию тромбоцитов. У большинства больных отмечаются появление продуктов распада фибрина в циркулирующей крови и тромбоцитопения. Наличие циркулирующих мономеров фибрина (указывает на продолжающееся внутрисосудистое свертывание) определяется у меньшинства больных; предполагается, что у большинства больных к моменту обращения за медицинской помощью внутрисосудистое свертывание прекращается. Фактически во всех случаях наблюдается отложение фибрина в сосудах почечных клубочков, что приводит к сужению или облитерации просвета капилляров, снижению скорости клубочковой фильтрации и уменьшению перфузии почечных канальцев с их вторичной дисфункцией или некрозом. У большинства больных с болезнью Гассера возникает тромбоцитопения, которая обусловлена как возросшим потреблением тромбоцитов, так и сокращением периода их жизни. Более того, циркулирующие в крови тромбоциты неполноценны; в них меньше серотонина, аденозиндифосфата, бета-тромбоглобулина и снижена способность к агрегации, что предполагает их истощение . Значение этих факторов и развитии поражений почек или ЦНС не установлено, однако при возникновении у больного геморрагических осложнений тромбоцитопении следует помнить о сниженной функции тромбоцитов.
Другим удивительным фактом является очевидное снижение продукции простациклина (PGJ2) эндотелиальными клетками у некоторых больных с болезнью Гассера и членов их семей. Простациклин является эндогенным ингибитором агрегации тромбоцитов. Это предполагает, что у некоторых людей имеется генетический дефект, который может привести к болезни Гассера, если этиологический фактор вызовет повреждение сосудов. Такой механизм может быть причиной заболевания в наблюдавшихся семейных случаях.
У больных с Гемолитико-уремическим синдромом при снижении гематокрита до 10-15 % быстро возникает гемолитическая анемия. Реакция Кумбса, как правило, бывает отрицательной, а осмотическая резистентность и ферменты эритроцитов находятся в пределах нормы. Таким образом, анемия у них обусловлена, вероятно, микроангиопатией, приводящей к повреждению эритроцитов во время их продвижения по измененной капиллярной сети. Выживаемость перелитых с кровью клеток также снижена. Главной причиной повреждения эритроцитов является механическая травма, чему в ряде случаев способствует измененный антиоксидантный статус эритроцитов.
Наконец, как показывают проведенные за последнее десятилетие исследования, при болезни Гассера в процесс вовлекаются и другие органы, особенно печень и мозг, а также сердце и легкие, возможно, вследствие все той же микроангиопатии, обсуждавшейся выше. Судороги, ступор или кома нередко бывают осложнением гемолитико-уремического синдрома, и их нельзя объяснить только гипертензией, вызванной почечной недостаточностью, или нарушением электролитного баланса. Тем не менее патологические изменения в мозге детей, умерших от болезни Гассера, были неспецифическими (отек, некроз клеток); микротромбы (предположительно вследствие повреждения сосудов) обнаружены лишь в нескольких случаях.
Продромальное заболевание обычно на несколько дней или недель предшествует болезни Гассера и чаще всего проявляется диареей и рвотой. Кровавый стул отмечается не всегда, но довольно часто. Реже заболеванию предшествует инфекция верхних или нижних дыхательных путей или вирусная инфекция . Затем, прежде чем синдром начнет проявляться, может последовать короткий период относительно хорошего состояния. Наблюдаемые симптомы варьируют в зависимости от тяжести заболевания и вовлечения в процесс различных органов. Часто при первом обращении к врачу, связанном с жалобами на бледность или вялость ребенка, обнаруживаются анемия и уремия. Гемолитико-уремический синдром может также проявляться абдоминальными симптомами, симулирующими острый живот или язвенный колит, с кровавой диареей и болезненностью брюшной стенки при надавливании.
Симптомы заболевания широко варьируют как при вне почечных, так и при почечных проявлениях. Различия в тяжести заболевания связаны скорее с экстраренальными проявлениями, нежели с выраженностью и продолжительностью почечной недостаточности. У нетяжелых больных наблюдаются анемия, тромбоцитопения и азотемия, у более тяжелых - олигурия или анурия и гипертензия. У самых тяжелых больных могут также отмечаться упорные судороги, кома, легочная недостаточность или признаки миокардита.
Дифференциальный диагноз болезни Гассера включает септицемию и неотложные абдоминальные хирургические состояния с сопутствующей преренальной олигурией или острым тубулярным некрозом (заворот кишок с инфарктом, непроходимость кишечника, перфорация кишечника, дивертикул подвздошной кишки, слизистая колика). однако в мазках крови при болезни Гассера обычно определяется гемолиз, а абдоминальные признаки, как правило, обнаруживают доброкачественный характер. Факторы тяжелого васкулита при почечной недостаточности редко выявляются в детском возрасте (например, люпус-нефрит) и обычно не вызывают тромбоцитопении.
Включает выявление наличия тромбоцитов в мазке и определение числа лейкоцитов, коагуляционного профиля, содержания электролитов, функции печени, измерение уровня азота мочевины в крови и креатинина, а также культуральные исследования крови и кала. Особое внимание следует уделить выявлению патогенных штаммов кишечной палочки, в частности 0157:Н7, которому приписывается ведущая роль: в патогенезе болезни Гассера . У детей со значительными нарушениями психики, судорогами или патологическими изменениями, обнаруженными при неврологическом исследовании, можно произвести компьютерную томографию черепа и поясничную пункцию для исключения кровотечения или менингита. Рутинная биопсия почки не показана, она проводится лишь в атипичных случаях и при необходимости инструментального исследования.
В настоящее время при болезни Гассера осуществляется поддерживающее лечение, направленное на сохранение гематокрита) в приемлемых пределах, на нормализацию содержания электролитов в сыворотке и поддержание водного баланса, а также на борьбу с гипертензией и судорогами. Раннее проведение перитонеального диализа для коррекции биохимических показателей сыворотки и восстановления объема крови снижает смертность, тяжелобольных с 77% до менее чем 10%.
Рутинный непрерывный мониторинг в отделении интенсивной терапии включает контроль за ЭКГ. Для контроля за внутрисосудистым объемом и серийного получения проб крови для лабораторных анализов устанавливают центральновенозный катетер У больных с лабильной (транзиторной) гипертензией целесообразно использование внутриартериального катетера. Катетер для выведения мочи позволяет выполнять точное количественное определение диуреза и клиренса креатинина. В случае анурии этот катетер удаляют ввиду риска инфицирования.
Количество жидкостей, не содержащих натрия или калия, ограничивают объемом, необходимым для возмещения неощутимых потерь воды (300 мл/м 2 в день) плюс ее потеря с мочой и фекалиями. Другие виды поддерживающего лечения, имеют своей целью контроль за гиперкалимией, гипертензией и судорогами, если они возникают. Гиперкалиемию, если она не является неотложным состоянием, можно лечить с помощью катионообменных смол; глюкоза, инсулин и хлорид кальция используются при неотложной терапии. При не значительно выраженной или умеренной гипертензии лечение начинают с диализа для уменьшения внутрисосудистого объема; если симптомы сохраняются, назначают сосудорасширяющие средства. Для предупреждения тахикардии и повышения эффективности лечения можно добавить бета - блокаторы, такие как анаприлин. Тяжелая гипертензия лучше всего лечится с помощью непрерывной инфузии нитропруссида натрия при постоянном АД-мониторинге с использованием внутриартериального катетера. При сохранении гипертензии после окончания острой фазы болезни можно перевести больного на пероральный прием гипотензивных средств.
Осуществляется ежедневный контроль за гематокритом и числом тромбоцитов. Если гематокритное число меньше 20 %, больному переливают эритроцитную массу. Необходимо иметь под рукой запас крови на случай внезапного снижения гематокритного числа в результате гемолиза или кровотечения. Переливание тромбоцитов производится при определении их числа ниже 20,0*10 9 /л или при наличии клинических признаков кровотечения.
Сообщалось о применении антикоагулянтной терапии гепарином или противотромбоцитарными средствами, а также фибринолитической терапии стрептокиназой, однако число проспективных контролируемых исследований их эффективности пока невелико. Антикоагулянтная терапия, не дает немедленного антитромботического эффекта, но способна оказывать продолжительное благоприятное влияние на гипертензию и протеинурию в тяжелых случаях при комбинированном назначении гепарина и стрептокиназы. Однако антикоагулянтная терапия сопряжена со значительным риском геморрагических осложнений; у большинства детей выздоровление наступает без ее применения.
Немногочисленные литературные данные не позволяют дать окончательную оценку эффективности плазмафереза при лечении болезни Гассера. Описано лишь два случая такого лечения; в одном из них уровень креатинина в сыворотке крови начал снижаться еще до проведения плазмафереза на 14-й день болезни. Оценивалась также эффективность повторного переливания плазмы при лечении болезни Гассера у 10 детей и 7 взрослых. Отсутствие данных о проведении исследований в контрольной группе затрудняет интерпретацию полученных результатов; в целом же исход болезни был не лучше обычно наблюдаемого при стандартном лечении. У 2 больных (13 %,) в результате переливания развился сывороточный гепатит. Степень риска по отношению к эффективности рутинного переливания плазмы, по-видимому, слишком высока, что не позволяет рекомендовать повторные переливания плазмы больным с неослож ненной болезнью Гассера. Более определенно установлена положительная роль плазмафереза или переливания плазмы в лечении специально отобранных больных с рецидивирующим гемолитико-уремическим синдромом или с явными признаками нарушения синтеза простациклина.
На основании существующих клинико-патологических корреляций можно выделить подгруппы форм гемолитико-уремического синдрома, что помогает врачу в выборе лечения и прогнозировании исхода. Классическая форма болезни Гассера, наблюдаемая у маленьких пациентов, проявляется преимуществвенно поражением почечных клубочков и имеет хороший прогноз (табл. 1).
Таблица 1. Прогноз ГУС при разных формах заболевания.
| Форма болезни | Возраст | Возможная этиология | Прогноз |
| Классическая | Литература: |
1. Интенсивная терапия в педиатрии . Под ред. Дж. П. Моррея, М. 1995.
2. Неотложные состояния в педиатрии . Под ред. В. М. Сидельникова, Киев 1994.
3. Emerging Infectious Diseases , Vol.1, No. 4 - October-December 1995, pages 134-140
Гемолитико-уремический синдром - острое патологическое состояние, характеризующееся одновременным развитием микроангиопатической гемолитической анемии, тромбоцитопении и азотемии. Гемолитико-уремический синдром может проявляться кровавой диареей, абдоминальными болями, бледностью и иктеричностью кожи и склер, пастозностью лица, петехиями на коже, анурией, поражением ЦНС, печени, поджелудочной железы и сердца. Диагноз гемолитико-уремического синдрома основан на характерных клинических признаках, результатах общего и биохимического анализа крови и мочи, коагулограммы, бакпосева кала. Лечение гемолитико-уремического синдрома включает патогенетическую, симптоматическую и заместительную терапию.
МКБ-10
Общие сведения
Гемолитико-уремический синдром (болезнь Гассера) – тяжелое полиэтиологическое расстройство, проявляющееся сочетанием неиммунной гемолитической анемии, тромбоцитопении и острой почечной недостаточности. Гемолитико-уремический синдром наблюдается преимущественно у детей грудного и младшего возраста (с 6 мес. до 4 лет), но также встречается у детей старшего возраста и редко у взрослых. Ежегодно в расчете на 100 тыс. детского населения регистрируются 2-3 случая гемолитико-уремического синдрома у детей до 5 лет и 1 случай у детей до 18 лет. Поскольку гемолитико-уремический синдром - одна из частых причин острой почечной недостаточности у детей, то от своевременности его диагностики и лечения зависит исход заболевания.
Причины
У детей частыми причинами гемолитико-уремического синдрома являются острая кишечная инфекция (90%) и инфекции верхних дыхательных путей (10 %).
Основное значение в развитии Д+ гемолитико-уремического синдрома имеет энтерогеморрагическая Е. coli, продуцирующая специфический шига-подобный веротоксин, способный избирательно повреждать эндотелиальные клетки сосудов почек и головного мозга. Наибольшее сродство веротоксина с эндотелием капилляров почек наблюдается у детей первых 3 лет жизни. Веротоксин вызывает эндотелиальный апоптоз и лейкоцитозависимое воспаление, а также запускает цепь патологических реакций, приводящих к гемолизу эритроцитов, агрегации и деструкции тромбоцитов, локальной активации процесса коагуляции и внутрисосудистого отложения фибрина, развитию ДВС-синдрома.
Такими же свойствами обладает шигатоксин S. dysenteriae I типа. Развивающиеся микроциркуляторные нарушения (микроангиопатическая гемолитическая анемия, тромбоцитопения и микротромбозы) приводят к ишемическим изменениям в органах мишенях. При гемолитико-уремическом синдроме на фоне ОКИ наиболее часто поражаются капилляры клубочков почек, что может приводить к снижению скорости гломерулярной фильтрации, ишемии или некрозу клубочков, вторичной дисфункции или некрозу почечных канальцев, при массивном поражении – к ОПН.
Заражение энтерогеморрагической Е. coli может произойти при контакте с животными (кошками, крупным рогатым скотом) или инфицированным человеком; употреблении недостаточно термически обработанных мясных изделий, непастеризованных молочных продуктов, фруктовых соков, загрязненной воды. Для гемолитико-уремического синдрома характерна сезонность: на фоне ОКИ - преимущественно теплое время года (июнь-сентябрь), на фоне вирусных инфекций - зимне-весенний период.
Д- гемолитико-уремический синдром может быть постинфекционным, лекарственным, поствакцинальным, наследственным, связанным с системными заболеваниями соединительной ткани, идиопатическим. В 40% случаев развитие Д- гемолитико-уремического синдрома обусловлено респираторной инфекцией, возбудителем которой является S. pneumoniae, разрушающий мембраны эритроцитов, тромбоцитов и эндотелиоцитов с помощью фермента нейраминидазы. Вирусы ветряной оспы, ВИЧ, гриппа, Эпштейна-Барра, Коксаки также могут быть причиной гемолитико-уремического синдрома.
Установлена связь между развитием гемолитико-уремического синдрома у взрослых и употреблением некоторых медикаментов (циклоспорина А, митомицина С, эстроген - содержащих контрацептивов, противоопухолевых препаратов), трансплантацией костного мозга, злокачественными новообразованиями, системной красной волчанкой и антифосфолипидным синдромом, беременностью. Выявлены семейные случаи гемолитико-уремического синдрома с аутосомным типом наследования обусловленные дефектом системы комплемента, нарушением обмена простациклина, недостаточностью антитромботических факторов и др.
В основе гемолитико-уремического синдрома может лежать активация тромбоцитов иммунными комплексами (например, комплексом антиген – антитело после прививок живыми вакцинами против полиомиелита, против ветряной оспы, против кори, АКДС).
Классификация
В зависимости от этиологии и клинических особенностей разделяют гемолитико-уремический синдром диареяассоциированный - Д+ (типичный) и не ассоциированный с диареей - Д- (спорадический или атипичный). Д+ гемолитико-уремический синдром чаще встречается у детей раннего и младшего возраста, является эндемическим (распространен в Поволжье, Московском регионе); недиарейный – более свойственен детям старшего возраста и взрослым.
По тяжести течения выделяют легкую и тяжелую формы гемолитико-уремического синдрома.
- Легкая форма гемолитико-уремического синдрома подразделяется на тип А (анемия, тромбоцитопения и азотемия) и тип Б (триада симптомов в сочетании с судорожным синдромом или артериальной гипертензией);
- Тяжелая форма – на тип А (триада симптомов в сочетании с анурией длительностью более суток) и тип Б (триада симптомов в сочетании с анурией, артериальной гипертензией и судорожным синдромом).
Симптомы гемолитико-уремического синдрома
В клинической картине гемолитико-уремического синдрома различают продромальный период, разгар заболевания и восстановительный период. Продолжительность продромального периода составляет от 2 до 7 суток. Для него характерно появление признаков поражения ЖКТ или дыхательных путей.
Гемолитико-уремический синдром на фоне ОКИ, вызванной энтеропатогенной Е. coli, имеет ярко выраженную симптоматику. Развиваются симптомы гастроэнтерита или колита (часто кровавая диарея), тошнота, рвота, абдоминальные боли, лихорадка. Постепенно общее состояния ребенка ухудшается, повышенная возбудимость сменяется вялостью.
В период разгара гемолитико-уремического синдрома превалируют проявления гемолитической анемии, тромбоцитопении и ОПН: бледность и иктеричность кожного покрова, склер и слизистых оболочек; пастозность век, голеней; кожный геморрагический синдром в виде петехий или экхимозов, иногда - носовые кровотечения, в тяжелых случаях - снижение диуреза (олигурия или анурия). Тяжесть и продолжительность дизурии зависит от степени и глубины повреждения почек.
Гемолитико-уремический синдром может проявляться полиорганной патологией: поражением ЦНС, печени, поджелудочной железы, сердца, артериальной гипертензией. В 50% случаев гемолитико-уремического синдрома наблюдаются неврологические нарушения: подергивания мышц, гиперрефлексия, децеребрационная ригидность, гемипарезы, судороги, ступор, кома (особенно выраженные у детей первых лет жизни). Выявляются гепатоспленомегалия, кардиомиопатия, тахикардия, аритмия.
Продолжительность гемолитико-уремического синдрома обычно составляет 1-2 недели, затем наступает стабилизация и в 70% случаев - постепенное восстановление нарушенных функций: улучшение выделения мочи, повышение уровня тромбоцитов, нормализация уровня гемоглобина. При тяжелом течении наступает либо летальный исход вследствие экстраренальных поражений, либо формирование ХПН.
Диагностика
Диагноз гемолитико-уремического синдрома основан на выявлении характерных клинических признаков, осложняющих течение ОКИ или ОРВИ: гемолитической анемии, тромбоцитопении, ДВС-синдрома, азотемии.
При гемолитико-уремическом синдроме в крови обнаруживаются анемия, анизоцитоз и полихроматофилия эритроцитов (наличие фрагментированных форм), присутствие свободного гемоглобина, снижение количества тромбоцитов, лейкоцитоз, умеренная непрямая гипербилирубинемия, возрастание уровня мочевины и креатинина, гипонатриемия, гиперкалиемия, ацидоз (в олигоанурической стадии ОПН), гипоальбуминемия.
Моча приобретает коричневато-ржавый цвет, в ней могут появиться фибриновые комки, отмечается гематурия, протеинурия, гемоглобинурия. У детей с ОКИ выполняют бактериологическое исследование кала на выявление штаммов энтеропатогенной Е. coli. При тяжелых неврологических нарушениях возможно проведение КТ головного мозга и люмбальной пункции для исключения кровотечения и менингита.
Дифференциальная диагностика гемолитико-уремического синдрома проводится с неотложными хирургическими состояниями (аппендицитом, кишечной непроходимостью, окклюзией мезентериальных сосудов, перфорацией кишечника, дивертикулом подвздошной кишки), ишемическим колитом, септицемией с ДВС-синдромом, вирусным или бактериальным гастроэнтеритом, тяжелой степенью дегидратации при кишечных токсикозах, тромботической тромбоцитопенией.
Лечение гемолитико-уремического синдрома
Лечение гемолитико-уремического синдрома определяется периодом развития заболевания и тяжестью поражения почечной ткани. Чем раньше ребенок с гемолитико-уремическим синдромом поступает в стационар, тем выше вероятность его успешного и полного излечения. Патогенетическая терапия включает нормализацию агрегатного состояния крови с использованием антиагрегантов, гепаринотерапии; улучшение микроциркуляции (трентал, эуфиллин); коррекцию антиоксидантного статуса (витамины А и Е).
При бактериальной этиологии гемолитико-уремического синдрома назначаются антибиотики широкого спектра действия; при инфекции, вызванной энтеропатогенной Е. coli, прием антибиотиков и препаратов, замедляющих моторику кишечника, не рекомендуется. При олигоанурии показана коррекция водно-электролитных расстройств, подавление реакций метаболического распада и инфекционного процесса. Для коррекции тяжелой анемии используется инфузия эритроцитарной массы.
В половине случаев типичного гемолитико-уремического синдрома необходимо раннее проведение заместительной терапии: обменного плазмафереза, перитонеального диализа или гемодиализа. Гемодиализ проводится ежедневно в течение всего олигоуремического периода. В случае развития терминальной стадии ХПН показана трансплантация почки.
Прогноз
Гемолитико-уремический синдром имеет серьезный прогноз, летальность у маленьких детей во время острой фазы заболевания составляет 3-5%, у 12% развивается терминальная ХПН, у 25% происходит снижение клубочковой фильтрации. Плохой прогноз имеют атипичные наследственные, аутоиммунные и связанные с беременностью формы гемолитико-уремического синдрома.
Классическая форма гемолитико-уремического синдрома у детей раннего возраста с преимущественным поражением почечных клубочков протекает более благоприятно. В случае Д+ гемолитико-уремического синдрома наблюдается лучший исход по сравнению с недиарейным синдромом, сопровождающимся частыми рецидивами и высокой летальностью.
Читайте также: