
Регуляция клеточной гибели кратко
Обновлено: 02.07.2024
Под редакцией профессора Б.А. Фролова.
Издание рекомендовано Учебно-методическим объединением по медицинскому и фармацевтическому образованию вузов России в качестве учебного пособия для студентов, обучающихся по специальностям 060101.65 (лечебное дело), 060103.65 (педиатрия), 060105.65 (медико-профилактическое дело), 060201.65 (стоматология) и 060301.65 (фармация).
Рецензенты: заслуженный деятель науки РБ, акад. РАЕН, зав. кафедрой патофизиологии Башкирского государственного медицинского университета, д.м.н., проф. Д.А. Еникеев; зав. кафедрой общей и клинической патологии: патологическая анатомия, патологическая физиология Самарского государственного медицинского университета, д.м.н., проф. Т.А. Федорина.
Пособие посвящено одной из центральных проблем патофизиологии, общей и клинической патологии – гибели клетки. Представления о феномене клеточной гибели и ее механизмах имеют универсальное общебиологической значение при изучении патологических процессов, играющих роль в генезе врожденных дефектов развития, старения организма, онкологических, аутоиммунных, нейродегенеративных и других заболеваний. Понимание этих механизмов составляет не только важное ключевое звено в фундаментальной подготовке специалистов, но и обеспечивает возможность разработки новых подходов к решению практических задач, связанных с диагностикой и лечением ряда нозологий.
Настоящее пособие призвано содействовать восприятию феноменологических аспектов различных видов клеточной гибели: некробиотической, апоптотической, аутофагической, а также осмыслению биологической сущности изучаемых явлений.
Пособие содержит 5 глав, первая из которых посвящена основным понятиям, цитологическим и биохимическим критериям, используемым для характеристики клеточной гибели; этиологическим факторам, определяющим развитие апоптоза, некроза и аутофагии; сравнительной оценки их морфофункциональных проявлений.
Во второй главе рассматриваются свободнорадикальные механизмы некробиотической гибели клеток. Приводятся классификация, обозначаются источники и пути образования свободных радикалов в клетках человека и животных, в том числе радикалов кислорода и других его активных форм (АФК). Обсуждается участие АФК в регуляции физиологических процессов в клетках и в патогенезе повреждений компонентов клеточных структур: липидов, белков и молекул ДНК при оксидативном стрессе. Анализируются механизмы регуляции редокс-баланса клетки и значение факторов антиоксидантной защиты в его обеспечении как важнейшего условия повышения устойчивости клетки к различным агрессивным воздействиям окружающей среды и к нарушениям собственного внутриклеточного гомеостаза. Подчеркивается многоуровневый характер действия механизмов антиоксидантной защиты, отличающийся их сопряженностью и взаимокомпенсаторностью.
Глава четыре посвящена апоптозу. Обсуждаются биохимические механизмы программированной клеточной гибели, включая участие в ней поли-АДФ-рибозил-полимеразы (ПАРП) и каспаз. Приводятся классификация каспаз и сведения об их роли в апоптотическом демонтаже клеток. Представлены сигнальные каскады запуска апоптоза с включением внутреннего (митохондриального) и внешнего (рецепторно-опосредованного) путей. Дается оценка проапоптотической роли цитохрома С, его участию в формировании апоптосомы; приводятся сведения о про- и антиапоптотических протеинах семейства Bcl-2; рассматривается значимость свободных радикалов и белка р53 в индукции митохондриального пути запуска апоптоза. Представлена последовательность образования на основе гомотипических белок-белковых взаимодействий многокомпонентных сигнальных комплексов, участвующих в инициации рецепторно-опосредованного апоптоза с участием Fas/APO-1 и ТНФ R1-рецепторов. Рассматриваются механизмы контроля апоптотической гибели, направленные на блокировку сигнала с клеточных рецепторов, стерическое препятствование их гомотипической спонтанной агрегации, а также на ингибирование активности каспаз. Отдельно обсуждаются особенности перфорин-зависимого апоптоза и его роль в реализации клеточной цитотоксичности. Заключительный раздел главы включает анализ последствий нарушений апоптоза как при избыточной, так и при недостаточной выраженности этого процесса.
Материал пятой главы содержит сведения об аутофагической гибели клеток. Рассмотрены различные формы аутофагии (микроаутофагия, макроаутофагия и шаперон-опосредованная аутофагия), механизмы регуляции, взаимоотношения аутофагии с другими видами клеточной гибели: апоптотической и некробиотической. Дается оценка биологической роли аутофагии не только как одной из форм программированной гибели клеток, но и как механизму противодействия старению организма, а также как механизму адаптации к широкому спектру различных неблагоприятных воздействий. Приводятся сведения о последствиях нарушения аутофагии в формировании патологии, включая нейродегенеративные заболевания, сердечную недостаточность, наследственные миопатии, стеатоз печени и др. Обсуждается возможность лекарственной регуляции аутофагии.
Пособие адресовано студентам и аспирантам медицинских институтов, медицинских и биологических факультетов университетов, педагогических, сельскохозяйственных, спортивных учебных заведений, а также физиологам, врачам и преподавателям медико-биологических дисциплин.

Клеточная смерть — это необратимая дегенерация жизненно важных клеточных функций (синтез АТФ и поддержание окислительно-восстановительного гомеостаза), приводящая к потере целостности клетки (нарушение проницаемости цитоплазматической мембраны или фрагментация клетки). Данный процесс отличен от клеточного старения — необратимой потери пролиферативного потенциала, связанной с определенными морфологическими и биохимическими признаками, что сопровождается секрецией множества факторов (то есть клетка приобретает так называемый секреторный фенотип, ассоциированный со старением — SASP); при этом клеточное старение не является формой гибели клеток.

Рис.1. Хронология терминов, используемых в исследовании клеточной смерти [1].
Современная система классификации гибели клеток была обновлена Комитетом номенклатуры по гибели клеток (Nomenclature Committee on Cell Death, NCCD) в 2018 году, с учетом данных многолетней истории изучения этого загадочного процесса (рис. 1) [1]. Помимо фундаментального деления на случайную (accidental cell death, ACD) и регулируемую (regulated cell death, RCD) клеточную смерть, в последней дополнительно выделено несколько подпрограмм, имеющих как четкое физиологическое значение (например, некроптоз и пироптоз, которые наблюдаются во время эмбрионального развития и при вирусных инфекциях), так и представленные клеточными реакциями на специфические токсины (ферроптоз, энтоз, нетоз, партанатоз, зависимая от лизосом и аутофагии гибель клеток, а также недавно описанные формы гибели клеток, вызываемые изменением pH среды (алкалиптоз) и активными формами кислорода, без вовлечения уже известных механизмов (оксеиптоз)). Регуляция гибели клеток проявляется в том, что конкретные генетические или фармакологически-индуцированные изменения способны как запустить, так и прервать деструкцию клеток [2].
Интересно, что регулируемая гибель клеток не является уникальной для многоклеточных форм жизни, где она имеет ключевую роль в поддержании гомеостаза организма как в физиологических, так и в патологических условиях, но также встречается (в упрощенном варианте) среди одноклеточных эукариот, образующих (по крайней мере, в течение части их жизненного цикла) колонии (несколько видов дрожжей и Dictyostelium discoideum), апоптозоподобный вариант клеточной гибели описан у организмов Leishmania spp. и даже у некоторых прокариот (например, Escherichia coli) [3].

Рис. 2. Основные подпрограммы регулируемой клеточной смерти | Клетки, подвергающиеся нерепарируемым нарушениям внутриклеточной или внеклеточной микросреды могут активировать один из многих каскадов сигнальной трансдукции, что в конечном итоге приводит к их гибели. Каждый из таких режимов регулируемой гибели клеток инициируется и осуществляется молекулярными механизмами. Морфологические признаки вариабельны: от полностью некротического морфотипа до полностью апоптотического; иммуномодулирующее действие также различно: от противовоспалительного и толерогенного до провоспалительного и иммуногенного [2].
КС ПМП-клеточная смерть, регулируемая переходом митохондриальный проницаемости, осуществляется за счет формирования одноименной поры (также называемой мегапорой), в образовании и дальнейшем поддержании которой играет важную роль циклофилин D (CypD / пептидилпрол-цис-транс-изомераза F) на фоне снижения АТФ и повышения уровня кальция в клетке [5]. Сокращения: РКС-регулируемая клеточная смерть; АЗКС-аутофагия-зависимая клеточная смерть; ЛЗКС-лизома-зависимая клеточная смерть; ИКС - иммуногенная клеточная смерть.
В классификации сформулированы определения различных программ клеточной смерти.
Случайная гибель клеток — фактически мгновенная и неконтролируемая (собственно умирающей клеткой) форма гибели клеток, морфологически соответствующая деструкции и фрагментации компонентов цитоплазматической мембраны, вызванная экстремальными физическими, химическими или механическими воздействиями.
Регулируемая гибель клеток — форма гибели клеток, которая возникает в результате активации одного или нескольких модулей сигнальной трансдукции и, следовательно, может быть фармакологически или генетически смодулирована. Субпрограммы регулируемой гибели клеток описаны в табл. 1 [1, 2, 4].
Таблица 1

Список литературы:
1. Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery
of regulated cell death. Cell Res. 2019 (5):347-364.
2. Galluzzi L. et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018 (25): 486–541.
3. Pandey S. S., Singh S., Pathak C., Tiwari B. S. Programmed Cell Death: A Process of Death for Survival - How Far Terminology Pertinent for Cell Death in Unicellular Organisms. Journal of cell death. 2018 (11): 1179066018790259.
4. Singh, R., Letai, A., Sarosiek, K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019 (20): 175–193.


39 километров кишечника
Любая популяция клеток регулируется тремя процессами, одинаково важными: деление, дифференцировка — превращение молодых клеток в зрелые (при этом их количество может как увеличиваться, так и уменьшаться) и гибель клеток. Тело взрослого человека состоит из десятков триллионов клеток, и ежедневно каждый из нас теряет десятки миллиардов из них, в пересчете на вес — примерно килограмм. Само собой разумеется, потерю восполняют новые клетки, так что мы не теряем по килограмму в день. (Кстати, жировые клетки, которым желают погибели многие худеющие, с возрастом могут прибавляться в числе, а умирают неохотно.) Все мы знаем, как слущивается и обновляется поверхностный слой кожи — эпидермис. В числе наиболее активно гибнущих — клетки эпителия кишечника: на протяжении жизни человека они заменяются примерно 4000 раз. Если бы старые клетки не погибали, то за 70 лет наш кишечник достиг бы длины 39 км! Активно обновляются и клетки костного мозга — за те же 70 лет организм производит их около трех тонн. Еще один пример — тимус, в котором рождаются и созревают клетки иммунной системы. Примерно 90% тимоцитов — так называют лимфоциты, пока они находятся в тимусе, — в нем же и погибают, и лишь 10% выходит за его пределы.
В индивидуальном развитии человека или любого другого существа тоже не обойтись без программируемой гибели клеток. Хрестоматийный пример апоптоза — исчезновение хвоста у головастика; интересно, что этот процесс вместе с другими метаморфозами регулируется изменением уровня тироидного гормона в крови. А чтобы у животного сформировались пальцы на лапе, должны исчезнуть клетки, расположенные между зачатками пальцев (рис. 1). Программируемая гибель клеток участвует и в созревании половых органов, и в развитии мозга. Погибшие при апоптозе клетки организма должны быть съедены соседними клетками либо макрофагами — профессионалами пожирания. Благодаря этому апоптоз почти никогда не сопровождается воспалением. Подробнее об этом можно прочитать в недавно опубликованных статьях (H. Yamaguchi et al., 2014, Apoptosis: Keeping inflammation at bay, eLIFE, 3:e02172; D. Wallach, A. Kovalenko, 2014, Apoptosis: Keeping inflammation at bay, eLIFE, 3:e02583).
Рис. 1. Апоптоз во время нормального развития конечности мыши. Клетки, подвергшиеся апоптозу, ярко окрашены. Справа — та же конечность день спустя (W. Wood et al., “Development”, 2000, 127:5245–5252)
Очевидно, что смерть клеток должна строго регулироваться, они должны погибать в определенное время и в определенном месте, иначе в организме воцарится хаос.
Бабочка и червь
Тогда же, в 60-е, биолог Сидней Бреннер, выходец из Южной Африки, работавший в Великобритании, предложил новый модельный объект для исследования индивидуального развития организмов — червячка Caenorhabditis elegans, обитающего в почве. Эти крошечные существа интересны тем, что тела взрослых особей состоят из строго определенного числа клеток, участь каждой из которых предопределена. Через четыре десятилетия, в 2002 году, Сидней Бреннер вместе с Робертом Хорвитцом и Джоном Салстоном получили Нобелевскую премию по физиологии или медицине за идентификацию генов нематоды, контролирующих развитие органов и программируемую гибель клеток.
Пересадки на путях гибели
Механизмы регуляции клеточной смерти оказались весьма сложными, и, несмотря на колоссальный прогресс в этой области, многое остается непонятным. Необходимо детально разобраться в сигнальных путях, приводящих к гибели клетки. Сейчас считается, что существует основной, сердцевинный (коровый) путь с ответвлениями, которые ведут или к специфическим механизмам гибели клеток в отдельных тканях, или к патологиям.
Номенклатурный комитет по исследованию гибели клеток, в который я имею честь входить, по совокупности морфологических и биохимических изменений выделил четыре типичных вида клеточной смерти — апоптоз, некроз, аутофагию и корнификацию (ороговение), а также восемь атипичных видов. Каждый из них протекает по своему пути. При этом нельзя сказать, что типичные важнее атипичных, они просто лучше изучены.
Несколько лет назад Европейский союз выделил 12 миллионов евро на поддержку исследовательского проекта, в котором участвовали биологи- экспериментаторы, врачи, специалисты по математическому моделированию из 12 стран. Мне посчастливилось руководить этим проектом. Его задачей было исследовать сигнальные пути, ведущие к апоптозу и другим типам гибели клеток при ВИЧ-инфекции и онкологических заболеваниях, в частности раке легких, а также в нормальных клетках. (Выбор пал на эти заболевания не только из-за их большой значимости: при СПИДе наблюдается избыточная гибель клеток, при раке — недостаточная.) Эксперименты проводили на клеточных культурах человека, на модельных организмах — дрожжах, нематоде C. elegans и мышах, выполняли также клинические испытания. Проект завершился в 2013 году; в результате удалось получить тесты для выявления патологий и разработать подходы к новым методам терапии.
Что касается теоретического выхода — итоговая схема путей клеточной гибели (рис. 2) довольно сложна, и рассказать о ней полностью в короткой статье невозможно. И все же нетрудно заметить, что перспективных мишеней для воздействия довольно много (на схеме они обозначены минусами). На этих этапах клеточную смерть можно остановить или, если заменить минусы плюсами, ускорить.

Рис. 2. Схема путей, по которым сигнал апоптотической гибели доходит до митохондрий и дальше распространяется по клетке, заканчиваясь распадом хроматина и фрагментацией ядра. Важную роль в прохождении апоптотического сигнала играет активация каспаз, семейства протеолитических ферментов.
Hsp — белки теплового шока, Cyt. c — цитохром c; подробнее о каспазах, белках семейств Bcl-2, IAP, а также SMAC, иначе называемом Diablo, см. в тексте
Одна из сложностей состоит в том, что белки, участвующие в регуляции гибели клеток, выполняют и другие функции. Это и понятно: трудно представить, чтобы рациональная природа создала специальную систему исключительно для умерщвления клеток. По логике вещей, составляющие этой системы должны в норме делать какую-нибудь полезную работу, а при необходимости мобилизоваться, чтобы убрать патологические клетки. Такая многофункциональность усложняет терапию: воздействуя на звено апоптозного пути, важно не помешать работе этого компонента в нормальной ткани.
Почетное место в апоптотической форме клеточной гибели занимают каспазы — семейство из тринадцати белков, разделенных на две группы, которые участвуют в развитии апоптоза или воспаления. Каспазы относятся к протеазам — ферментам, расщепляющим другие белки, причем результаты этой их активности могут быть самыми разными, даже когда речь идет об одном и том же ферменте, но в разных тканях и при различных условиях. Так, при окислительном стрессе каспаза 1 расщепляет интерлейкин 1В, превращая его в активную форму. (Интерлейкины играют центральную роль в иммунных и воспалительных процессах.) Это может быть причиной ишемии в клетках печени и миокарда; на клеточном уровне происходит апоптоз, который в случае нарушений фагоцитоза может трансформироваться в некроз. В печеночной ткани та же каспаза может расщепить белок, приводя к переключению апоптотической программы в аутофагическую, а затем и к геморрагическому шоку. С другой стороны, если полностью убрать этот белок, это вызывает гибель клеток печени по типу некроза.
В 1990–2000-е годы многие фармацевтические фирмы вкладывали огромные деньги в разработку ингибиторов каспаз. Теперь практически все прекратили работу в этом направлении, поскольку ингибиторы оказались токсичными, — именно потому, что блокируют нормальную функцию каспаз в клетках. В настоящее время ингибиторы каспаз используют лишь в экстренных ситуациях, например при остром циррозе печени, когда необходимо как можно скорее остановить разрушение ткани. Другой пример — такое тяжелое заболевание, как болезнь Крона: хроническое воспаление всех отделов желудочно-кишечного тракта, от полости рта до прямой кишки, с образованием свищей, инфекционными осложнениями и прочими проблемами. При лечении болезни Крона (а также ревматоидного артрита и язвенного колита) хорошо показал себя препарат инфликсимаб, в России известный как ремикейд, — он действует как раз через каспазу-1.
Белки семейства IAP — ingibitors of apoptosis proteases — в соответствии с названием, ингибируют апоптотические протеазы, то есть каспазы, тем самым выключая апоптоз. В нормальных клетках белки IAP может обезвредить митохондриальный белок SMAC (second mitochondria-derived activator of caspases) — он выходит из митохондрий, соединяется с IAP и убирает их функцию. Логично было использовать этот эффект для терапии. И действительно, низкомолекулярные миметики SMAC (небольшие молекулы, имитирующие функцию этого белка) показали себя достаточно эффективными при терапии глиомы — опухоли мозга (рис. 3). По некоторым обмолвкам врачей в российских СМИ можно предположить, что подобными препаратами (но, конечно, не только ими) лечили в США певицу Жанну Фриске.
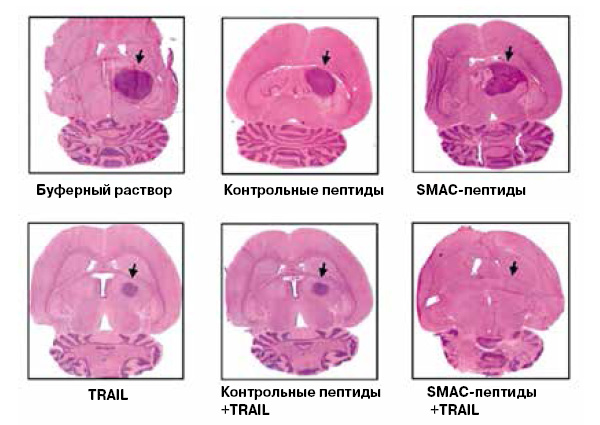
10.1038/nm735). В настоящее время данные миметики SMAC находятся на третьей фазе клинических испытаний" border=0 >
Рис. 3. Подобные SMAC вещества (пептиды или низкомолекулярные органические соединения) делают злокачественную опухоль мозга — глиому чувствительной к терапии белком TRAIL, цитокином из семейства факторов некроза опухолей, а также к многим химиотерапевтическим препаратам (не указаны на рисунке). Только при их совместном действии глиома в мозге подопытных мышей исчезает полностью, ее клетки гибнут по пути апоптоза (S.Fulda et al, Nature Medicine, 2002, 8 (8), 808–815, DOI:10.1038/nm735). В настоящее время данные миметики SMAC находятся на третьей фазе клинических испытаний
Следующий важный элемент схемы — Bcl-2. Перенос его гена с одной хромосомы на другую (транслокация) ассоциируется с лимфомой В-клеток. Отсюда название белка и его гена — аббревиатура B cell lymphoma. В 80-е годы ХХ века австралийский биолог Дэвид Во с коллегами показал, что этот белок работает как антиапоптотический, препятствуя гибели В-клеток; вскоре это подтвердили и другие исследователи. Таким образом, впервые было доказано, что белки, участвующие в негативной регуляции гибели клеток, могут работать как онкогены: если апоптоз блокирован и дефектные клетки не погибают, заболевание развивается.
С этой публикацией связана интересная история. Дэвид Во в то время был аспирантом в Институте медицинских исследований Уолтера и Элизы Холл в Мельбурне. Его научный руководитель, доктор Сьюзен Кори, результаты Дэвида по Bcl-2 встретила холодно. Но Дэвид, будучи упорным человеком, отправился за поддержкой к своему второму руководителю — доктору Джерри Адамсу, и тот решил, что работа заслуживает внимания. Интрига заключалась в том, что второй руководитель был мужем первого. Итогом рабочих и, возможно, внерабочих дискуссий стала совместная публикация руководителей и аспиранта (D. L. Vaux, S. Cory, J. M. Adams, Nature, 1988, 335, 440–442).
Интересно, что аутофагия в опухоли может как подавлять ее развитие, так и способствовать ему. Однако совокупность последних данных говорит о том, что можно заставить аутофагию работать только на гибель опухоли. Не исключено, что удастся как-то использовать связь между аутофагией и апоптозом, переключения между этими двумя маршрутами.
Возможности аутофагии в борьбе с онкологическими заболеваниями наши лаборатории в МГУ и в Каролинском институте изучают совместно с клиницистами из Российского онкологического научного центра имени Н. Н. Блохина. Идея выглядела парадоксально: не стимулировать, а подавить аутофагию в клетках опухоли. Известно, что при этом в клетке накапливаются активные формы кислорода (АФК) и она становится более чувствительной к инициации процесса гибели. Мы попытались проверить это на практике и убедились, что идея работает: ингибирование аутофагии на определенных участках привело к накоплению АФК, и, если в этот момент подействовать специфическими противоопухолевыми препаратами, можно эффективно убить опухоль. Замечу, что эта работа была выполнена только на аденокарциноме легкого, мы не проверяли результаты ни на каких других видах новообразований, и наше представление о механизме пока остается рабочей гипотезой.
Характерный пример — рак легкого. Это название объединяет по крайней мере четыре разных заболевания: мелкоклеточный и немелкоклеточный рак, который, в свою очередь, делится еще на три вида: аденокарциному, плоскоклеточный и крупноклеточный рак. Это деление отнюдь не формальное: у них абсолютно разные генетические основы, биохимия, этиология, общего — только локализация в легком. Конечно же и лечить их нельзя одинаково.
Нужно учитывать еще и такой фактор, как индивидуальная чувствительность больных к терапии. Около 15 лет назад в США был создан препарат для лечения аденокарциномы и других немелкоклеточных раков, получивший название Иресса (гефитиниб). Испытания на клетках в культуре и на животных показали хорошие результаты, и, поскольку рак легких очень распространен в Японии, американское Агентство по контролю пищевых продуктов и лекарств (FDA) решило проводить третью фазу клинических испытаний именно там. Примерно треть пациентов с аденокарциномой легкого отвечала на терапию — великолепное достижение. Но когда FDA допустило этот препарат к применению в США, произошло фиаско: эффект был всего у 2% больных. Дело в том, что Иресса — ингибитор рецептора эпидермального фактора роста EGF, известного как онкоген, а при аденокарциноме могут быть мутации в гене этого белка. В Японии определенная мутация, ранее не известная, встречалась у 30% пациентов, а в Америке приблизительно у 2% — им-то и помогал препарат. Не случайно Евросоюз поддерживает сейчас большую программу персональной медицины. Программа весьма дорогостоящая, но без нее не продвинуться вперед.
Делящиеся и неделящиеся клетки. Митоз. Дифференцировка и специализация клеток. Этапы жизненного цикла специализированной клетки. Некроз и апоптоз. Регуляция численности клеток в организме.
До сих пор много тайн клетки остаются неразгаданными. Загадочным во многом остается и запрограммированный генетически алгоритм ее жизни, названный жизненным циклом клетки (клеточным циклом). Жизненный цикл клетки (рисунок 1.3.14) начинается с момента ее образования после деления родительской клетки и заканчивается либо новым делением, либо превращением в специализированную клетку.
Рисунок 1.3.14. Жизненный цикл клетки:
1 - интерфаза; 2 - митоз; 3 - дифференцировка; 4 - функционирование специализированной клетки
Большинство клеток продолжает делиться. Им свойственен клеточный цикл, состоящий из периодически повторяющихся стадий: так называемой интерфазы (1) – этапа подготовки к делению и непосредственно процесса деления – митоза (2). К этапам дифференцировки (3) и функционирования специализированной клетки (4) мы вернемся чуть позже.
На стадии подготовки к делению происходит удвоение генетического материала (редупликация ДНК). Масса клетки во время интерфазы увеличивается до тех пор, пока она примерно вдвое не превысит начальную. Отметим, что сам процесс деления намного короче этапа подготовки к нему: митоз занимает примерно 1/10 часть клеточного цикла.
Цикличность (периодическое повторение) стадий интерфазы и митоза можно проиллюстрировать на примере фибробластов – одного из видов клеток соединительной ткани (рисунок 1.3.15). Так, нормальные фибробласты эмбриона человека размножаются приблизительно 50 раз. Каков генетически запрограммированный предел возможных делений клетки – это одна из неразгаданных тайн биологии.
Рисунок 1.3.15. Цикличность стадий интерфазы и митоза:
1 - интерфаза, стадия подготовки к митозу; 2 - митоз (деление клетки)
Хотя все клетки появляются путем деления предшествующей (материнской) клетки (“Всякая клетка от клетки”), не все они продолжают делиться. Клетки, достигшие некоторой стадии развития при дифференцировке, могут терять способность к делению.
Дифференцировка – возникновение различий в процессе развития первоначально одинаковых клеток, приводящее к их специализации. Процесс дифференцировки заключается в последовательном считывании и использовании наследственной информации, что обеспечивает синтез различных белков (в первую очередь ферментов), характерных для данного вида клеток. Другими словами, различия между клетками определяются набором белков, синтезируемых в клетках определенного вида.
При дифференцировке набор хромосом в клетке не меняется, изменяется лишь соотношение активных и неактивных генов, кодирующих различные белки.
Существуют два типа регуляции экспрессии (активации или блокирования) генов:
- Кратковременная адаптивная активация (реже блокирование), зависящая, в частности, от концентрации вещества, включающегося в обмен веществ (исходного вещества или продукта метаболизма). Этот механизм выработался эволюционно как приспособительная реакция и особенно ярко проявляется у животных (например, быстрый синтез пигментов у хамелеона в зависимости от условий).
- Длительное (в течение всей жизни клетки и/или многих генераций клеток!) блокирование или активация гена, возникающее в ходе клеточной дифференцировки. Например, в ДНК любой клетки желудка есть ген, отвечающий за синтез белков, из которых состоит ноготь. Но он необратимо блокирован гистонами и другими белками (этот участок ДНК плотно упакован), что никогда не позволит считывать с него информацию. Поэтому в желудке не растут ногти; а гены, ответственные за синтез гемоглобина, функционируют только у молодых форм эритроцитов, но не действуют в зрелых эритроцитах или других клетках.
На рисунке 1.3.14 цифрами 3 и 4 отмечены этапы дифференцировки и активного функционирования специализированной клетки.
Нервные клетки мозга, однажды возникнув, уже не делятся. В течение жизни число нейронов постепенно уменьшается. Поврежденные ткани мозга неспособны восстанавливаться путем регенерации. Однако изначально число нейронов в мозге настолько велико, что до конца жизни человека они способны поддерживать необходимые связи в нервной системе.
В качестве примера клеток, неспособных к делению, можно рассмотреть эритроциты. Как известно, эритроциты в процессе специализации теряют ядро, следовательно, не имеют в своем составе ДНК. Возникают эритроциты из так называемой стволовой клетки костного мозга. Клеткой-предшественницей (стволовой клеткой) называют клетки кроветворной ткани, которые на протяжении всей жизни человека сохраняют способность делиться и, тем самым, поставлять дочерние клетки, которые в дальнейшем будут специализироваться в одном направлении и замещать погибшие клетки. Срок жизни и активного функционирования эритроцитов невелик (около 4 месяцев), затем они разрушаются, в основном в селезенке.
Этапы жизни специализированной клетки, неспособной к делению (нейрона, эритроцита), условно можно изобразить на оси времени линией, разделенной на несколько отрезков (рисунок 1.3.16). Эти отрезки дают представление о временном соотношении периодов жизни такой клетки: рождения, созревания и активного функционирования, угасания (старения) и естественной гибели.
Рисунок 1.3.16. Этапы жизненного цикла специализированной клетки:
1 - рождение в процессе деления материнской клетки; 2 - созревание и дифференцировка; 3 - активное функционирование; 4 - угасание (старение); 5 - запрограммированная клеточная гибель
Время протекания каждого этапа и продолжительность жизненного цикла для однотипных клеток в нормальных условиях практически одинаковы.
Например, эритроциты живут 90-125 дней, а тромбоциты – всего 4 суток. Это говорит о том, что клетки используют для отсчета времени своей жизни некий механизм, алгоритм, заложенный в них природой. И в каждый момент жизни клетка строго следует законам, продиктованным этим алгоритмом.
На всех этапах клеточного цикла варьируют значения некоторых параметров жизнедеятельности клетки, и, в частности, отмечается различная скорость и интенсивность протекания процессов метаболизма (рисунок 1.3.17). Это обусловлено, в первую очередь, непрерывно меняющейся активностью ферментов, благодаря которым протекают все реакции в клетке. Ферменты могут синтезироваться в клетке “по мере надобности”, активироваться, временно блокироваться или полностью разрушаться (подробнее о ферментах будет сказано в разделе 1.4.3).
Рисунок 1.3.17. Интенсивность метаболизма на различных этапах жизни клетки:
1 - рождение; 2 - созревание и дифференцировка; 3 - активное функционирование; 4 - угасание (старение); 5 - запрограммированная клеточная гибель
Рассмотрим подробнее наиболее характерные процессы, происходящие на каждом из этапов клеточного цикла.
Рождение. Отправным моментом жизни любой клетки (кроме половой, для которой характерен мейоз) считают деление материнской клетки с образованием двух идентичных дочерних – митоз (от греческого mitos – нить). Во время митоза основная задача материнской клетки – поровну передать равноценный в количественном и качественном отношении генетический материал дочерним клеткам.
Митоз часто называют “танцем хромосом”. Каждая следующая фигура в этом танце не случайна, здесь нет ни одного лишнего или бессмысленного “па” – это еще один четкий, выверенный природой алгоритм. В. Дудинцев в романе “Белые одежды” так описывает процесс деления клетки: “Хромосомы шевелились, как клубок серых червей, потом вдруг выстроились в строгий вертикальный порядок. Вдруг удвоились – теперь это были пары. Тут же какая-то сила потащила эти пары врозь, хромосомы подчинились, обмякли, и что-то повлекло их к двум разным полюсам.”
Деление клетки на две идентичные (митоз) характеризуется сменой нескольких морфологически и физиологически различающихся стадий (рисунок 1.3.18). На первой стадии митоза хроматин плотно упаковывается (этот процесс называется суперспирализацией хроматина) с образованием хромосом (1). Каждая хромосома состоит из двух идентичных половинок (хроматид) – будущих дочерних хромосом. Затем при сокращении так называемого веретена деления (2), представляющего собой комплекс микротрубочек и микрофибрилл, дочерние хромосомы расходятся, буквально подтягиваются нитями веретена деления к противоположным полюсам клетки. После окончательного расхождения дочерние хромосомы вновь раскручиваются, превращаясь в длинные и тонкие нити хроматина (3). Веретено деления исчезает, хроматин в дочерних клетках окружается ядерной оболочкой, и между дочерними клетками образуется поперечная перетяжка (4) из клеточных мембран.
Рисунок 1.3.18. Последовательность стадий митоза (схема):
1 - хромосомы; 2 - веретено деления; 3 - хроматин; 4 - поперечная перетяжка
Хромосомы, как мы уже говорили, представляют собой максимально плотно упакованные нити ДНК, с которых на этапе деления невозможно считывание информации. Соответственно, на этапе деления не происходит биосинтеза белка, интенсивность процессов метаболизма минимальна, транспорт веществ в клетку и из нее практически равен нулю. Все процессы в делящейся клетке направлены на выполнение главнейшей задачи – максимально точно, без искажения, передать генетическую информацию дочерним клеткам, – в ущерб второстепенным (на данном этапе!) функциям.
Созревание. В этот период происходит дифференцировка клеток и становление ключевых ферментных систем. Клетка готовится выполнять предназначенные природой функции, постепенно активизируя свой обмен веществ.
Активное функционирование. Интенсивность реакций метаболизма и сопряженного с ним энергетического обмена в это время максимальны.
Процессы в клетке направлены на обеспечение постоянства внутренней среды и выполнение специфических функций: нейрон воспринимает и передает нервный импульс, эритроцит переносит кислород и так далее.
Угасание (старение). Этот процесс запрограммирован генетически и, в первую очередь, проявляется уменьшением выработки и активности ферментов в клетке. При этом замедляются биохимические реакции, тормозится метаболизм и энергетический обмен.
Стареющие клетки, как правило, имеют неудвоенное количество ДНК, но сохраняют жизнеспособность и некоторую метаболическую активность в течение определенного времени.
Естественная гибель клетки (апоптоз). К сожалению, до сих пор процесс естественной гибели клеток до конца не изучен.
Известно, что в клетке из-за блокирования ферментов прекращается синтез белка, а нет белка – нет и жизни. Морфологически апоптоз характеризуется разрушением ядра и цитоплазмы. “Осколки” погибшей клетки поглощаются и перерабатываются специальными клетками иммунной системы – фагоцитами. Но ведь клетки могут погибнуть и под воздействием случайных факторов (механических, химических и любых других). Случайная гибель клеток (а также ткани, органа) в биологии называется некрозом. Важно то, что естественная клеточная гибель (апоптоз) в отличие от некроза не вызывает воспаления в окружающих тканях.
В организме запрограммированная клеточная гибель выполняет функцию, противоположную митозу, и, тем самым, регулирует общее число клеток в организме. Апоптоз играет важную роль в защите организма при вирусных инфекциях. В частности, иммунодефицит при ВИЧ-инфекции определяется нарушениями в контроле апоптоза.
Теперь, когда мы рассмотрели все этапы жизненного цикла клеток, коротко остановимся на процессах регуляции численности клеток в организме. Во время эмбриогенеза (первого этапа внутриутробного развития) число клеток постоянно возрастает, причем в геометрической прогрессии (рисунок 1.3.19).
Рисунок 1.3.19. Увеличение численности клеток на этапе эмбриогенеза
Зигота, образовавшаяся после слияния яйцеклетки и сперматозоида, делится с образованием двух дочерних клеток. Затем, в результате последовательных делений, образуются четыре, восемь, шестнадцать клеток и так далее. Параллельно с увеличением численности на этапе эмбриогенеза происходит дифференцировка клеток – так образуются ткани (смотри раздел 1.5.1).
Во взрослом организме общая численность клеток стабильна, она остается практически неизменной на протяжении многих лет (рисунок 1.3.20).
Рисунок 1.3.20. Поддержание постоянства общей численности клеток во взрослом организме
Это происходит за счет уравновешивания процессов возникновения новых клеток (митоза) и гибели клеток, естественной (апоптоза) или случайной (некроза). При смещении равновесия, например, гибели большого количества клеток в результате травмы или другого негативного воздействия, включаются механизмы регенерации (увеличение интенсивности деления клеток для замещения погибших), о которых уже было сказано. Таким образом, общая численность клеток поддерживается практически на постоянном уровне.
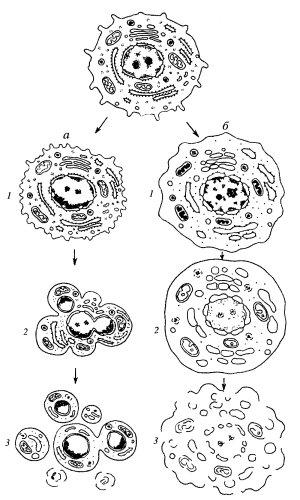
Рис. 354. Два пути клеточной гибели
а — апоптоз (программированная клеточная смерть): 1 — специфическое сжатие клетки и конденсация хроматина, 2 — фрагментация ядра, 3 — фрагментация тела клетки на ряд апоптических телец;
6 — некроз: 1 — набухание клетки, вакуолярных компонентов, конденсация хроматина (кариорексис), 2 — дальнейшее набухание мембранных органоидов, лизис хроматина ядра (кариолизис), 3 — разрыв мембранных компонентов клетки - лизис клетки
Этот вид клеточной смерти обычно связывается с нарушением внутриклеточного гомеостаза в результате нарушения проницаемости клеточных мембран, приводящим к изменению концентрации ионов в клетке, с необратимыми изменениями митохондрий, что сразу приводит к прекращению всех жизненных функций, включая синтез макромолекул. Некроз вызывают повреждения плазматической мембраны, подавление активности мембранных насосов под действием многих ядов, а также необратимые изменения энергетики при недостатке кислорода (при ишемии происходит закупорка кровеносного сосуда) или отравлении митохондриальных ферментов (действие цианидов). При этом при повышении проницаемости плазматической мембраны клетка набухает за счет ее обводнения, в цитоплазме происходит увеличение концентрации ионов Na + и Са 2+ , закисление цитоплазмы, набухание вакуолярных компонентов и разрыв их мембран, прекращение синтеза белков в цитозоле, освобождение лизосомных гидролаз и лизис клетки. Одновременно с этими изменениями в цитоплазме изменяются и клеточные ядра: вначале они компактизируются (пикноз ядер), но по мере набухания ядра и разрыва его оболочки пограничный слой хроматина распадается на мелкие массы (кариорексис), а затем наступает кариолизис - растворение ядра. Особенностью некроза является то, что такой гибели подвергаются большие группы клеток (например, при инфаркте миокарда из-за прекращения снабжения кислородом участка сердечной мышцы). Обычным является то, что участок некроза подвергается атаке лейкоцитов и в зоне некроза развивается воспалительная реакция (см. рис. 354).
Важно отметить, что во всех случаях апоптоза — во время ли эмбрионального развития, во взрослом ли организме, в норме или при патологических процессах — морфология процесса гибели клеток очень сходна. Это может говорить об общности процессов апоптоза в разных организмах и в разных органах.
Исследования на разных объектах показали, что апоптоз есть результат реализации генетически запрограммированной клеточной гибели. Первые доказательства наличия генетической программы клеточной смерти (ПКС) были получены при изучении развития нематоды Caenorhabditis elegans. Этот червь развивается всего за трое суток, и его малые размеры позволяют проследить за судьбой всех его клеток, начиная с ранних этапов дробления до половозрелого организма.
Оказалось, что при развитии С. elegans образуется всего 1090 клеток, из которых часть нервных клеток в количестве 131 штуки спонтанно погибает путем апоптоза и в организме остается 959 клеток. Были обнаружены мутанты, у которых процесс элиминации 131 клетки был нарушен. Были выявлены два гена ced-З и ced-4, продукты которых вызывают апоптоз 131 клетки. Если у мутантных C. elegans эти гены отсутствуют или изменены, то апоптоз не наступает и взрослый организм состоит из 1090 клеток. Был найден и другой ген - ced-9, который является супрессором апоптоза: при мутации ced-9 все 1090 клеток погибают. Аналог этого гена был обнаружен у человека: ген bcl-2 также является супрессором апоптоза различных клеток. Оказалось, что оба белка, кодируемые этими генами, — Ced-9 и Всl-2, имеют один трансмембранный домен и локализуются во внешней мембране митохондрий, ядер и эндоплазматического ретикулума.
Система развития апоптоза оказалась очень сходной у нематоды и позвоночных животных, она состоит из трех звеньев: регулятора, адаптера и эффектора. У C. elegans регулятором является Ced-9, который блокирует адаптерный белок Ced-4, который в свою очередь не активирует эффекторный белок Ced-З, протеазу, которая действует на белки цитоскелета и ядра (табл. 16).
Развитие процесса программированной клеточной смерти (апоптоза)

У позвоночных система ПКС более сложная. Здесь регулятором является белок Всl-2, который ингибирует адаптерный белок Араf-1, стимулирующий каскад активации специальных протеиназ — каспаз.
Каспазы — цистеиновые протеазы, которые расщепляют белки по аспарагиновой кислоте. В клетке каспазы синтезируются в форме латентных предшественников — прокаспаз. Существуют инициирующие и эффекторные каспазы. Инициирующие каспазы активируют латентные формы эффекторных каспаз. Субстратами для действия активированных каспаз служат более 60 различных белков. Это, например, киназа фокальных адгезионных структур, инактивация которой приводит к отделению апоптических клеток от соседей; это ламины, которые при действии каспаз разбираются; это цитоскелетные белки (промежуточные филаменты, актин, гельзолин), инактивация которых приводит к изменению формы клетки и к появлению на ее поверхности пузырей, которые дают начало апоптическим тельцам; это активируемая протеаза САD, которая расщепляет ДНК на олигонуклеотидные нуклеосомные фрагменты; это ферменты репарации ДНК, подавление которых предотвращает восстановление структуры ДНК, и многие другие.
Одним из примеров разворачивания апоптозного ответа может являться реакция клетки на отсутствие сигнала от необходимого трофического фактора, например фактора роста нервов (NGF), или андрогена (рис. 355).

Рис. 355. Одна из моделей развития апоптоза при отсутствии трофического фактора
1 — плазматическая мембрана; 2 — внешняя мембрана митохондрии; 3 —трофический фактор; 4 — рецептор трофического фактора; 5 — дефосфорилирование проапоптозного белка Bad; 6 — белок Bad инактивирует антиапоптозный белок Bcl-2; 7 — выход цитохрома с в цитозоль; 8 — активация апоптозного белка Вах, открывание ионных каналов; 9 — цитохром с активирует адаптерный белок Apaf-1; 10 — активация прокаспазы 9; 11 — активация каспазы 9; 12 — активация каспазы 3; 13 — конденсация и деградация хроматина; 14 —деградация ядерной ламины; 15 —деградация цитоскелета
В цитоплазме клеток в присутствии трофических факторов находится в неактивной форме еще один участник реакции — фосфорилированный белок Bad. В отсутствие трофического фактора этот белок дефосфорилируется и связывается с белком Всl-2 на внешней митохондриальной мембране и этим ингибирует его антиапоптозные свойства. После этого активируется мембранный проапоптический белок Вах, открывая путь ионам, входящим в митохондрию. В это же время из митохондрий через образовавшиеся в мембране поры в цитоплазму выходит цитохром с, который связывается с адаптерным белком Apaf-1, который в свою очередь активирует прокаспазу 9. Активированная каспаза 9 запускает каскад других прокаспаз, в том числе каспазу 3, которые, будучи протеиназами, начинают переваривать мешенные белки (ламины, белки цитоскелета и др.), что вызывает апоптическую смерть клетки, ее распад на части, на апоптические тельца.
Апоптические тельца, окруженные плазматической мембраной разрушенной клетки, привлекают отдельные макрофаги, которые их поглощают и переваривают с помощью своих лизосом. Макрофаги не реагируют на соседние нормальные клетки, но узнают апоптические. Это связано с тем, что при апоптозе нарушается асимметрия плазматической мембраны и на ее поверхности появляется фосфатидилсерин, негативно заряженный фосфолипид, который в норме располагается в цитозольной части билипидной плазматической мембраны. Таким образом, путем избирательного фагоцитоза ткани как бы очищаются от погибших апоптозных клеток.
Как указывалось выше, апоптоз может быть вызван целым рядом внешних факторов, таких как радиация, действие некоторых токсинов, ингибиторов клеточного метаболизма. Необратимые повреждения ДНК вызывают апоптоз. Это связано с тем, что накапливающийся транскрипционный фактор — белок р53, не только активирует белок р21, который ингибирует зависящую от циклина киназу и останавливает клеточный цикл в G1- или G2-фазе (см. рис. 353), но и активирует экспрессию гена Ьах, продукт которого запускает апоптоз.
Избирательные повреждения митохондрий, при которых в цитоплазму высвобождается цитохром с, также являются частой причиной развития апоптоза. Особенно митохондрии и другие клеточные компоненты страдают при образовании токсически активных форм кислорода (АТК), под действием которых во внутренней мембране митохондрий образуются неспецифические каналы с высокой проницаемостью для ионов, в результате чего матрикс митохондрий набухает, а внешняя мембрана разрывается. При этом растворенные в межмембранном пространстве белки вместе с цитохромом с выходят в цитоплазму. Среди освободившихся белков есть факторы, активирующие апоптоз, и прокаспаза 9.
Многие токсины (рицин, дифтерийный токсин и др.), а также антиметаболиты могут вызывать гибель клеток путем апоптоза. При нарушении синтеза белка в эндоплазматическом ретикулуме в развитии апоптоза участвует локализованная там прокаспаза 12, которая активирует ряд других каспаз, и в том числе каспазу 3.
Элиминация — удаление отдельных клеток путем апоптоза, наблюдается и у растений. Здесь апоптоз включает в себя, так же как у животных клеток, фазу индукции, эффекторную фазу и фазу деградации. Морфология гибели клеток растений сходна с изменениями клеток животных: конденсация хроматина и фрагментация ядра, олигонуклеотидная деградация ДНК, сжатие протопласта, его дробление на везикулы, разрыв плазмодесм и т.д. Однако везикулы протопласта разрушаются гидролазами самих везикул, так как у растений нет клеток, аналогичных фагоцитам. Так, ПКС происходит при росте клеток корневого чехлика, при формировании перфораций у листьев, при образовании ксилемы и флоэмы. Опадание листьев связано с избирательной гибелью клеток определенной зоны черенка.
Биологическая роль апоптоза, или программированной смерти клеток, очень велика: это удаление отработавших свое или ненужных на данном этапе развития клеток, а также удаление измененных или патологических клеток, особенно мутантных или зараженных вирусами.
Итак, для того чтобы клетки в многоклеточном организме существовали, нужны сигналы на их выживание — трофические факторы, сигнальные молекулы. Эти сигналы могут быть переданы на расстояние и уловлены соответствующими рецепторными молекулами на клетках-мишенях (гормональная, эндокринная сигнализация), это может быть паракринная связь, когда сигнал передается на соседнюю клетку (например, передача нейромедиатора). При отсутствии таких трофических факторов реализуется программа апоптоза. В то же время апоптоз может вызываться сигнальными молекулами, например при резорбции хвоста головастиков под действием тироксина. Кроме того, действие ряда токсинов, влияющих на отдельные звенья метаболизма клетки, также может стать причиной клеточной гибели посредством апоптоза.
Читайте также: