
Особенности клинических проявлений наследственной патологии кратко
Обновлено: 03.07.2024
Врожденные заболевания являются одной из распространенных причин младенческой и детской смертности, развития хронических болезней у детей и инвалидизации.
Быстрый переход
Врожденные заболевания могут быть обусловлены генетическими мутациями (передающимися по наследству или спонтанными), инфекциями матери во время беременности (цитомегаловирус, ветряная оспа, краснуха), воздействием лекарств и химических веществ, загрязняющих воздух, воду или пищу. Причины множества врожденных дефектов до сих пор неизвестны.
Генетическими или наследственными факторами обусловлены около 20 % врожденных заболеваний. К ним относятся нарушения, при которых мутация затрагивает один ген (серповидно-клеточная анемия); хромосомные нарушения, при которых хромосомы (или их части) отсутствуют (синдром Тернера) или имеют структурные изменения (увеличение количества хромосом или трисомия при синдроме Дауна); многофакторные нарушения, вызванные мутациями двух и более генов. Врожденные дефекты и нарушения развития могут вызывать делеции или дупликации отдельных генов (изменения митохондриальной ДНК). Примером такого заболевания является муковисцидоз, характеризующееся поражением экзокринных желез и жизненно-важных органов (легких и желудочно-кишечного тракта). Врожденные заболевания также ассоциированы со случайными (новыми) повреждениями генов, спонтанными (не наследующимися от родителей) мутациями (большинство случаев ахондроплазии).
По статистике, врожденные заболевания имеют 2–3 % младенцев. К возрасту 1 года их число увеличивается до 5 %, поскольку не все эти патологии диагностируются сразу после рождения.
Врожденные (наследственные) заболевания также классифицируют по типу наследования. При аутосомно-доминантном наследовании заболевание может передаваться от родителя к ребенку в 50 % случаев (мышечная дистрофия Дюшенна, хорея Гентингтона). При аутосомно-рецессивном наследовании генетическая аномалия передается ребенку только в том случае, если оба родителя наделены одним и тем же дефектным геном (муковисцидоз, серповидно-клеточная анемия). Здесь частота наследования составит 25 %, то есть в среднем у 1 ребенка из 4 детей этих родителей будет аутосомно-рецессивное заболевание.
Патологические состояния могут передаваться и при наследовании, сцепленном с полом (наследование гена, находящегося в половых хромосомах). Х-сцепленные рецессивные заболевания почти всегда ограничены мужским полом (гемофилия, дальтонизм, мышечная дистрофия Дюшенна). Х-сцепленные доминантные заболевания встречаются как у мальчиков, так и у девочек, однако у детей мужского пола имеют более тяжелое течение (Х-сцепленный гипофосфатемический рахит). Y-сцепленные заболевания встречаются довольно редко, поскольку Y-хромосома содержит всего несколько генов (ихтиоз).
Некоторые врожденные заболевания формально не относятся к генетическим, но имеют ту или иную выраженность наследования: наследуются факторы риска либо сам ген, но с низкой пенетрантностью (частотой проявления гена в признаках).
Гемофилия
Гемофилия — группа редких наследственных нарушений свертываемости крови, вызванных дефицитом необходимого белка (фактора свертывания крови).
Гемофилия A (классическая) встречается чаще (>80 % случаев) и связана с дефицитом VIII фактора свертывания крови, гемофилия B (болезнь Кристмаса) — реже (>10 % случаев), она обусловлена недостаточностью IX фактора свертывания крови. Гемофилия C встречается очень редко, обусловлена дефицитом XI фактора свертывания крови, чаще всего в классификации группы ее не упоминают.
Заболевание относится к X-сцепленным с полом, наследуется по рецессивному признаку по женской линии. Классическую гемофилию вызывают мутации гена F8, расположенного на X-хромосоме. Примерно в 70 % случаев заболевание наследуется по Х-сцепленному образцу, в остальных случаях оно возникает спонтанно (новая мутация), в дальнейшем спонтанное заболевание становится наследственным. Гемофилией A болеют практически исключительно мужчины, редкие случаи заболевания у женщин, носительниц дефектного гена, почти всегда характеризуются легким течением. Гемофилию B вызывают мутации гена F9, так же расположенного на X-хромосоме, она характерна для мужчин, женщины болеют очень редко. В некоторых случаях заболевание возникает спонтанно (приобретенная гемофилия A или B) и тоже связано с недостаточностью VIII или IX факторов свертывания крови. Приобретенная гемофилия является аутоиммунным заболеванием, при котором организм вырабатывает антитела, атакующие факторы свертывания крови (чаще всего VIII фактор). Примерно в половине случаев приобретенной гемофилии у пациента имеется связанное с ней основное состояние или заболевание (беременность, аутоиммунные заболевания, миелопролиферативные заболевания, воспалительные заболевания кишечника и др.), в остальных эпизодах причина остается невыясненной.
Гемофилия A затрагивает примерно 1 из 5 000 новорожденных мальчиков, гемофилия B — примерно 1 из 25 000. Около 60 % пациентов имеют тяжелую форму гемофилии, обычно им ставят диагноз при рождении или в течение первых 2 лет жизни.
Возраст манифестации гемофилии и тяжесть течения заболевания зависят от уровня активности факторов свертывания крови. Легкая форма характеризуется уровнем фактора свертывания крови (VIII или IX), превышающим 5 % от нормы, средняя — 1–5 %, тяжелая — ниже 1 %. У большинства пациентов, независимо от тяжести течения заболевания, эпизоды кровотечения чаще встречаются в раннем детском, детском и подростковом возрасте, чем в дальнейшей взрослой жизни.
При легкой и умеренной формах гемофилии длительные кровотечения могут возникнуть только в результате травмы, хирургического вмешательства или стоматологической процедуры. Нередко диагноз гемофилии ребенку устанавливают к 5–6 годам, обратив внимание на длительное посттравматическое кровотечение, длительное кровотечение во время стоматологического лечения или операции. К другим заметным симптомам легкого и умеренного течения гемофилии относятся непроходящие гематомы (синяки), частые носовые кровотечения, кровоточивость десен.
Для тяжелой формы гемофилии характерны эпизоды спонтанного кровотечения, которые приводят к кровоизлияниям различной локализации — в мягкие ткани, мышцы, суставы. При гемартрозах (кровоизлияние в полость сустава) возникает ограничение подвижности суставов, сопровождающееся острой болью и воспалением. У пациентов с тяжелой формой гемофилии спонтанные кровотечения чаще всего происходят в мышцы и суставы, однако могут затрагивать любой внутренний орган, включая почки, органы ЖКТ, головной мозг (гематурия, мелена, гематохезия, желудочно-кишечные кровотечения, внутричерепные кровотечения). Тяжелая форма гемофилии A обычно проявляется в раннем детском возрасте, диагноз чаще всего устанавливается к 2 годам ребенка. Обычными симптомами (при отсутствии лечения заболевания) здесь являются кровотечения из-за незначительных травм ротовой полости (прикусывание губ, языка, щек), подкожные гематомы, большие шишки после удара головой, спонтанные кровотечения (2–5 эпизодов в месяц).
Диагноз гемофилии A или B устанавливается на основании симптомов, истории личного и семейного анамнеза пациента, лабораторных исследований (общего анализа крови, коагулограммы, оценивающей состояние системы свертывания крови и активности ее белков, молекулярно-генетического исследования). Парам из группы риска по рождению ребенка с гемофилией необходимо получить генетическую консультацию на этапе планирования беременности. Определение конкретной мутации гена F8 (или F9) помогает не только выявить женщин-носительниц дефектного гена в конкретной семье, но и полезно для пренатальной диагностики гемофилии (амниоцентез, биопсия хориона).
Некоторым пациентам с легкой формой гемофилии назначается синтетический аналог вазопрессина — десмопрессин, повышающий VIII фактор свертывания крови (вводится внутривенно или интраназально), а также антифибринолитические средства, замедляющие распад факторов свертывания крови.
Примерно в 30 % случаев лечения тяжелых случаев гемофилии длительная заместитетельная терапия может приводить к изоиммунизации — образованию антител к вводимым факторам свертывания крови (иммунная система распознает вводимый фактор VIII как чужеродный). Этот процесс может сопровождаться аллергическими реакциями (разной степени тяжести), возрастает риск жизнеугрожающих кровотечений. В этом случае пациенту назначается альтернативное лечение — плазмаферез, иммунодепрессанты.
Гемохроматозы
Гемохроматоз — наследственное заболевание, характеризующееся нарушением обмена железа и его накоплением в тканях и органах.
Заболевание сопровождается повышенным всасыванием железа в желудочно-кишечном тракте и накоплением в печени, сердце, поджелудочной железе, суставах, гипофизе, что приводит к полиорганной недостаточности и развитию таких болезней, как цирроз печени, рак печени, диабет, болезни сердца и суставов.
Наследственный гемохроматоз вызывают мутации генов HFE, HFE2, HAMP, SLC40A1 и TfR2, он классифицируется в зависимости от возраста начала, генетической причины и способа наследования: гемохроматоз 1 типа, гемохроматоз 2 типа, гемохроматоз 3 типа, гемохроматоз 4 типа. Вторичный гемохроматоз ассоциирован с другими заболеваниями (не является наследственным) — анемией, хронической болезнью печени, инфекциями.
Гемохроматоз, редко возникающий в младенческом возрасте и не имеющий явной причины, называют неонатальным гемохроматозом. При этой форме заболевания избыточное накопление железа в тканях и органах начинается еще до рождения ребенка. Неонатальный гемохроматоз быстро прогрессирует и характеризуется поражениями печени, которые выявляют при рождении или в первые дни жизни. Дети с этим заболеванием часто рождаются недоношенными или имеют нарушения внутриутробного развития. Точная причина неонатального гемохроматоза неизвестна, есть версия, что он развивается в том случае, если иммунная система матери распознает клетки печени ребенка как чужеродные. Симптомы обычно включают гипогликемию, нарушения свертываемости крови, пожелтение кожных покровов и склер глаз, отеки. Диагноз устанавливается на основании признаков и симптомов, лабораторных и инструментальных исследований, биопсии печени. Лечение включает переливание крови, внутривенное введение иммуноглобулинов, трансплантацию печени.
Гемохроматоз 1 типа ассоциирован с мутациями в гене HFE, расположенном на коротком плече 6–й хромосомы, наследуется по аутосомно-рецессивному типу, является наиболее распространенным типом наследственного гемохроматоза и поражает в основном мужчин. Вероятность того, что ребенок унаследует мутацию в гене HFE от родителя составляет 25 %, вероятность того, что он станет носителем дефектного гена — 50 %.
Начальные симптомы заболевания обычно отмечаются в возрасте 40–60 лет и включают боль в животе, снижение полового влечения, усталость, слабость, боли в суставах, сухость кожи. В дальнейшем проявляются такие симптомы и осложнения, как изменение пигментации кожи (бронзовая кожа), выпадение волос на голове и туловище, аритмия, кардиомиопатия, хроническая сердечная недостаточность, сахарный диабет, гепатомегалия, цирроз печени, спленомегалия, атрофия яичек, аменорея (у женщин) и другие.
Диагноз устанавливается на основании симптомов заболевания, лабораторных исследований (уровень железа, ферритина, трансферрина, железосвязывающей способности сыворотки крови, маркеры функции печени, уровень глюкозы крови и др.), инструментальных исследований (рентгенография суставов, электрокардиография и эхокардиография сердца, УЗИ органов брюшной полости, МРТ печени и др.), биопсии печени, молекулярно-генетического исследования (в том числе для выявления родственников — имеющих гемохроматоз или носителей).
Лечение гемохроматоза направлено на удаление избытка железа из организма (флеботомия, хелатирование) с помощью забора крови (как при взятии анализов или донорстве крови, только в большем объеме) и специальных препаратов, образующих комплексное соединение с железом и способствующих его удалению из железосодержащих белков (дефероксамин).
Терапевтическая флеботомия сначала проводится 1–2 раза в неделю, поддерживающая — каждые 2–4 месяца. Вместе с этим проводится симптоматическое лечение сахарного диабета, патологий сердца, цирроза печени и других заболеваний, вызванных гемохроматозом. Пациентам запрещен прием препаратов железа и любых медикаментозных средств или комплексов, которые могут содержать железо и витамин C, запрещен алкоголь во избежание дальнейшего повреждения печени (если оно имеется), рекомендуется придерживаться диеты, исключающей продукты с высоким содержанием железа (красное мясо, яблоки, печень, шпинат).
Гемохроматоз 2 типа вызывают мутации в генах HFE2 или HAMP, он наследуется по аутосомно-рецессивному типу и проявляется чаще всего в детском возрасте. Для этого типа заболевания характерны следующие симптомы: высокие уровни ферритина и трансферрина, врожденный фиброз печени, артропатия, кардиомиопатия, генерализованная гиперпигментация кожи, мышечная слабость, гипогонадизм, задержка полового созревания, сахарный диабет, цирроз печени, остеопороз, спленомегалия, гепатомегалия и другие. Диагностика и лечение гемохроматоза 2 типа проводятся аналогично диагностике и лечению гемохроматоза 1 типа.
Гемохроматоз 3 типа вызывают мутации в гене TFR2, он наследуется по аутосомно-рецессивному типу. Симптомы обычно проявляются до 30 лет и, помимо вышеперечисленных, могут включать снижение уровня лимфоцитов и нейтрофилов в крови, красные или пурпурные пятна на коже. Диагностика и лечение гемохроматоза 3 типа проводятся аналогично диагностике и лечению гемохроматоза 1 и 2 типов.
Симптомы гемохроматоза 4 типа могут проявиться как в детском, так и во взрослом возрасте. Этот тип заболевания вызывают мутации гена SLC40A1, наследуется оно по аутосомно-доминатному типу. Вероятность того, что ребенок унаследует мутацию в гене SLC40A1 от родителя составляет 50 %, соответственно, вероятность того, что он станет носителем дефектного гена — тоже 50 %. Симптомы гемохроматоза 4 типа могут проявляться в виде повышенной утомляемости, слабости, болей в суставах, изменения пигментации кожи, затруднения дыхания, сердечной недостаточности, сахарного диабета, анемии, врожденного фиброза печени, цирроза печени, остеоартроза, катаракты. Диагностика и лечение гемохроматоза 4 типа проводятся аналогично диагностике и лечению гемохроматоза 1, 2 и 3 типов.
Наследственный гемохроматоз — заболевание с пониженной пенетрантностью (вероятностью того, что ген будет иметь любые фенотипические проявления) дефектного гена, т. е. у некоторых людей с мутациями гена HFE никогда не проявятся симптомы, при этом у их детей или других членов семьи с мутацией гена произойдет манифестация заболевания.
Мышечная дистрофия Дюшенна
Мышечная дистрофия Дюшенна — редкое наследственное заболевание, характеризующееся нарастающей мышечной слабостью с последующей атрофией мышц. Ассоциировано с мутацией гена DMD, расположенного на половой X-хромосоме (X-сцепленный рецессивный тип наследования).
Прежде чем обсуждать особенности проявления наследственной патологии, следует уточнить понятия врожденной, семейной и наследственной патологии.
Семейные болезни – все виды патологии, проявляющиеся у нескольких членов одной семьи (за исключением случаев острых инфекционных заболеваний). Семейные болезни могут иметь наследственную или ненаследственную природу, быть врожденными или приобретенными.
Наследственными являются все патологические состояния и болезни человека, которые обусловлены нарушениями в его генетической программе (генными и хромосомными мутациями), независимо от того, проявляются они при рождении или в процессе жизни, встречаются у отдельных членов семьи или нескольких родственников.
В основе особенностей клинической картины наследственной патологии лежат генетические закономерности действия и взаимодействия генов, закономерности их реализации в определенных условиях внешней по отношению к гену среды.
К числу особенностей наследственной патологии относятся: врожденный и семейный характер, хроническое, прогредиентное, рецидивирующее течение, полисистемность и множественность поражения, специфический характер и возрастозависимая манифестация патологических проявлений, отсутствие эффективных способов терапии, приуроченность ряда патологических состояний к определенным этническим группам населения.
1. Врожденный характер заболевания.К моменту рождения каждый организм обладает индивидуальным набором нормальных и патологических генов (аллелей), которые, реализуясь в определенных условиях среды, обуславливают состояние здоровья или наследственной патологии. Изменения генетической информации, составляющие основу наследственных болезней, могут быть унаследованы от родителей, либо возникают в генетическом аппарате половых клеток родителей, в зиготе или на ранних стадиях развития эмбриона. Поэтому все наследственные признаки человека (за исключением проявлений соматических мутаций) являются врожденными, независимо от времени их проявления, или манифестации.
2. Семейный характер заболевания. Большинство наследственных заболеваний проявляется поражением нескольких членов семьи, относящихся к одному или разным поколениям. Однако не все случаи семейной патологии являются наследственными. Например, профессиональные болезни в семьях-династиях, или инфекции, поражающие всех или многих контактных родственников. В то же время, наличие заболевания только у одного из членов семьи не исключает наследственного характера этой болезни, поскольку последняя может быть следствием спорадической мутации (хромосомные мутации или генные мутации, возникающие de novo) или гетерозиготности родителей по рецессивному аллелю (сегрегация мутантного фенотипа при фенилкетонурии). Кроме того, в силу неполной пенетрантности или низкой экспрессивности гена признаки болезни могут не проявляться даже у носителей патологического аллеля.
4. Хроническое, прогредиентное, рецидивирующее течение.Для наследственных болезней, начинающихся в любом возрасте, характерно длительное хроническое течение с прогредиентным (прогрессирующим), нередко рецидивирующим характером клинической картины. Например, легочная форма муковисцидоза всегда сопровождается хронической пневмонией с бронхоэктазами и периодически обостряется и осложняется присоединяющейся бактериальной или вирусной инфекцией. Хронический процесс при наследственных болезнях является следствием постоянного действия мутантного гена. Степень прогредиентности одного и того же заболевания различается у разных больных, что формально можно объяснить взаимодействием генов в генотипе (генотип каждого человека индивидуален). Рецидивирующий характер течения наследственной патологии, возможно, обусловлен не только особенностями функционирования генов у больных, но и воздействиями на них внешних и внутренних средовых факторов (нарушение питания, стресс, охлаждение, инфекции и т.д.).

5. Полисистемность и множественный характер пораженияотличает наследственные болезни от ненаследственных. При этом симптомы поражения разных органов и систем либо взаимосвязаны эмбриологически, физиологически, патогенетически и т.д., либо нет. Например, нарушение смыкания верхних дуг позвонков может сопровождаться или нет несмыканием кожи и/или нервной трубки на соответствующем уровне позвоночника. В результате этих эмбриологически связанных процессов могут формироваться: скрытая спинно-мозговая грыжа (spina bifida occulta), открытая спинно-мозговая грыжа (spina bifida aperta), содержащая разной степени деформированный спинной мозг и его оболочки (менингоцеле, менингомиелоцеле, менингомиелоцистоцеле), либо рахишиз, при котором спинной мозг как таковой отсутствует и имеет вид деформированной тонкой пластинки или желоба. Нередко при наследственных болезнях невозможно установить какой-либо связи между сопровождающими их симптомами. Так, при синдроме Лоуренса-Муна-Барде-Бидля невозможно установить каких-либо физиологических или эмбриологических связей между развивающимися у больных симптомами - ожирением, гипогенитализмом, преаксиальной полидактилией и пигментной дистрофией сетчатки. По мнению Э.Хадерна такая особенность наследственных болезней объясняется плейотропным, т.е. множественным, действием гена, при котором патологический ген проявляется независимо в зачатках разных тканей, и синдром формируется как сумма независимых эффектов. Следует подчеркнуть, что плейотропное проявление мутантного гена, обнаруживаемое при различных, если не при всех наследственных болезнях, не характерно для ненаследственной, в том числе мультифакториальной патологии, где в абсолютном большинстве случаев патологический процесс начинается локально, а затем уже может получить сколь угодно широкое развитие. Из сказанного следует, что на основании только описания клинической картины и сочетания определенных симптомов, можно выделять многие моногенные наследственные синдромы.
8. Аномалии и пороки развития внутренних органовнередко встречаются при тератогенных и наследственных синдромах и объясняются особенностями проявления мутантных генов на морфологическом уровне онтогенеза. Нарушение места и времени процессов морфогенеза (усиление или, наоборот, недостаточность пролиферации, дегенерации, миграции, адгезии и избирательной клеточной гибели) эмбриональных зачатков тканей и органов сопровождается формированием пороков и аномалий развития органов, недоразвитием или чрезмерным развитием отдельных частей тела, а иногда и всей его половины (так называемые синдромы гемигипертрофии).
9. Отсутствие эффекта от традиционного лечения –еще одна особенность наследственных болезней, особенно если лечение проводится в форме симптоматической и/или посиндромной терапии. Однако, если при наследственном заболевании расшифрованы ключевые звенья патогенеза и известен первичный биохимический дефект – есть возможность или, по крайней мере, надежда на разработку адекватной патогенетической терапии (в форме диетотерапии или специфической медикаментозной терапии). Такие способы лечения уже разработаны для ряда наследственных нарушений метаболизма (фенилкетонурия, галактоземия, целиакия, муковисцидоз, гепатолентикулярная дегенерация, болезнь Гоше и т.д.) и продолжают разрабатываться для других. Кроме того, в последние годы все более активно разрабатываются и внедряются в практическую медицину методы генотерапии, направленные на внедрение в клетку нормального варианта недостающего или аномально функционирующего гена.
10. Этническая приуроченность патологических признаков и состояний -характерная для ряда этнических групп населения встречаемость определенных, в первую очередь, аутосомно-рецессивных заболеваний. Например, высокая частота встречаемости болезни Альцгеймера и болезни Тея-Сакса в популяциях евреев-ашкенази, серповидно-клеточной анемии и талассемии в популяциях средиземноморского населения. Информированность врачей клиницистов по популяционным частотам и структуре наследственной патологии в определенных этнических группах населения способствует своевременной диагностике этих заболеваний. Каждая популяция должна знать особенности своего генофонда, спектр и частоту встречаемости характерных для ее населения наследственных заболеваний.
Знание всех описанных специфических особенностей семиотики наследственных болезней позволит клиницистам разного профиля правильно обследовать пациента, оценить наблюдаемую клиническую картину, своевременно предположить или исключить диагноз наследственного заболевания.
Прежде чем обсуждать особенности проявления наследственной патологии, следует уточнить понятия врожденной, семейной и наследственной патологии.
Семейные болезни – все виды патологии, проявляющиеся у нескольких членов одной семьи (за исключением случаев острых инфекционных заболеваний). Семейные болезни могут иметь наследственную или ненаследственную природу, быть врожденными или приобретенными.
Наследственными являются все патологические состояния и болезни человека, которые обусловлены нарушениями в его генетической программе (генными и хромосомными мутациями), независимо от того, проявляются они при рождении или в процессе жизни, встречаются у отдельных членов семьи или нескольких родственников.
В основе особенностей клинической картины наследственной патологии лежат генетические закономерности действия и взаимодействия генов, закономерности их реализации в определенных условиях внешней по отношению к гену среды.
К числу особенностей наследственной патологии относятся: врожденный и семейный характер, хроническое, прогредиентное, рецидивирующее течение, полисистемность и множественность поражения, специфический характер и возрастозависимая манифестация патологических проявлений, отсутствие эффективных способов терапии, приуроченность ряда патологических состояний к определенным этническим группам населения.
1. Врожденный характер заболевания.К моменту рождения каждый организм обладает индивидуальным набором нормальных и патологических генов (аллелей), которые, реализуясь в определенных условиях среды, обуславливают состояние здоровья или наследственной патологии. Изменения генетической информации, составляющие основу наследственных болезней, могут быть унаследованы от родителей, либо возникают в генетическом аппарате половых клеток родителей, в зиготе или на ранних стадиях развития эмбриона. Поэтому все наследственные признаки человека (за исключением проявлений соматических мутаций) являются врожденными, независимо от времени их проявления, или манифестации.
2. Семейный характер заболевания. Большинство наследственных заболеваний проявляется поражением нескольких членов семьи, относящихся к одному или разным поколениям. Однако не все случаи семейной патологии являются наследственными. Например, профессиональные болезни в семьях-династиях, или инфекции, поражающие всех или многих контактных родственников. В то же время, наличие заболевания только у одного из членов семьи не исключает наследственного характера этой болезни, поскольку последняя может быть следствием спорадической мутации (хромосомные мутации или генные мутации, возникающие de novo) или гетерозиготности родителей по рецессивному аллелю (сегрегация мутантного фенотипа при фенилкетонурии). Кроме того, в силу неполной пенетрантности или низкой экспрессивности гена признаки болезни могут не проявляться даже у носителей патологического аллеля.
4. Хроническое, прогредиентное, рецидивирующее течение.Для наследственных болезней, начинающихся в любом возрасте, характерно длительное хроническое течение с прогредиентным (прогрессирующим), нередко рецидивирующим характером клинической картины. Например, легочная форма муковисцидоза всегда сопровождается хронической пневмонией с бронхоэктазами и периодически обостряется и осложняется присоединяющейся бактериальной или вирусной инфекцией. Хронический процесс при наследственных болезнях является следствием постоянного действия мутантного гена. Степень прогредиентности одного и того же заболевания различается у разных больных, что формально можно объяснить взаимодействием генов в генотипе (генотип каждого человека индивидуален). Рецидивирующий характер течения наследственной патологии, возможно, обусловлен не только особенностями функционирования генов у больных, но и воздействиями на них внешних и внутренних средовых факторов (нарушение питания, стресс, охлаждение, инфекции и т.д.).
5. Полисистемность и множественный характер пораженияотличает наследственные болезни от ненаследственных. При этом симптомы поражения разных органов и систем либо взаимосвязаны эмбриологически, физиологически, патогенетически и т.д., либо нет. Например, нарушение смыкания верхних дуг позвонков может сопровождаться или нет несмыканием кожи и/или нервной трубки на соответствующем уровне позвоночника. В результате этих эмбриологически связанных процессов могут формироваться: скрытая спинно-мозговая грыжа (spina bifida occulta), открытая спинно-мозговая грыжа (spina bifida aperta), содержащая разной степени деформированный спинной мозг и его оболочки (менингоцеле, менингомиелоцеле, менингомиелоцистоцеле), либо рахишиз, при котором спинной мозг как таковой отсутствует и имеет вид деформированной тонкой пластинки или желоба. Нередко при наследственных болезнях невозможно установить какой-либо связи между сопровождающими их симптомами. Так, при синдроме Лоуренса-Муна-Барде-Бидля невозможно установить каких-либо физиологических или эмбриологических связей между развивающимися у больных симптомами - ожирением, гипогенитализмом, преаксиальной полидактилией и пигментной дистрофией сетчатки. По мнению Э.Хадерна такая особенность наследственных болезней объясняется плейотропным, т.е. множественным, действием гена, при котором патологический ген проявляется независимо в зачатках разных тканей, и синдром формируется как сумма независимых эффектов. Следует подчеркнуть, что плейотропное проявление мутантного гена, обнаруживаемое при различных, если не при всех наследственных болезнях, не характерно для ненаследственной, в том числе мультифакториальной патологии, где в абсолютном большинстве случаев патологический процесс начинается локально, а затем уже может получить сколь угодно широкое развитие. Из сказанного следует, что на основании только описания клинической картины и сочетания определенных симптомов, можно выделять многие моногенные наследственные синдромы.
8. Аномалии и пороки развития внутренних органовнередко встречаются при тератогенных и наследственных синдромах и объясняются особенностями проявления мутантных генов на морфологическом уровне онтогенеза. Нарушение места и времени процессов морфогенеза (усиление или, наоборот, недостаточность пролиферации, дегенерации, миграции, адгезии и избирательной клеточной гибели) эмбриональных зачатков тканей и органов сопровождается формированием пороков и аномалий развития органов, недоразвитием или чрезмерным развитием отдельных частей тела, а иногда и всей его половины (так называемые синдромы гемигипертрофии).
9. Отсутствие эффекта от традиционного лечения –еще одна особенность наследственных болезней, особенно если лечение проводится в форме симптоматической и/или посиндромной терапии. Однако, если при наследственном заболевании расшифрованы ключевые звенья патогенеза и известен первичный биохимический дефект – есть возможность или, по крайней мере, надежда на разработку адекватной патогенетической терапии (в форме диетотерапии или специфической медикаментозной терапии). Такие способы лечения уже разработаны для ряда наследственных нарушений метаболизма (фенилкетонурия, галактоземия, целиакия, муковисцидоз, гепатолентикулярная дегенерация, болезнь Гоше и т.д.) и продолжают разрабатываться для других. Кроме того, в последние годы все более активно разрабатываются и внедряются в практическую медицину методы генотерапии, направленные на внедрение в клетку нормального варианта недостающего или аномально функционирующего гена.
10. Этническая приуроченность патологических признаков и состояний -характерная для ряда этнических групп населения встречаемость определенных, в первую очередь, аутосомно-рецессивных заболеваний. Например, высокая частота встречаемости болезни Альцгеймера и болезни Тея-Сакса в популяциях евреев-ашкенази, серповидно-клеточной анемии и талассемии в популяциях средиземноморского населения. Информированность врачей клиницистов по популяционным частотам и структуре наследственной патологии в определенных этнических группах населения способствует своевременной диагностике этих заболеваний. Каждая популяция должна знать особенности своего генофонда, спектр и частоту встречаемости характерных для ее населения наследственных заболеваний.
Знание всех описанных специфических особенностей семиотики наследственных болезней позволит клиницистам разного профиля правильно обследовать пациента, оценить наблюдаемую клиническую картину, своевременно предположить или исключить диагноз наследственного заболевания.

Задачей медицинской (клинической) генетики является разработка методов диагностики, лечения и профилактики наследственных болезней человека. К настоящему времени описано свыше 3500 наследственных болезней, около 5-5,5 % детей рождаются с наследственной или врожденной патологией.
Наследственные факторы и факторы среды
С генетической точки зрения все болезни в зависимости от роли наследственных и средовых факторов в их развитии можно подразделить на 3 группы.
- Наследственные заболевания. Фенотипическое проявление мутации как этиологического фактора практически не зависит от среды; последняя может только изменять выраженность симптомов и тяжесть течения болезни. Это генные и хромосомные наследственные болезни (гемофилия, фенилкетонурия, муковисцидоз, болезнь Дауна и др.).
- Болезни с наследственной предрасположенностью. Их в свою очередь можно подразделить еще на два вида. Болезни, наследственность при которых является этиологическим фактором, но для их проявления необходимо действие соответствующего фактора внешней среды (например, подагра, диабет).
Болезни, этиологическими факторами при которых являются средовые влияния, однако частота возникновения и тяжесть течения болезней зависят от наследственной предрасположенности. К таким болезням относятся атеросклероз, гипертоническая болезнь, язвенная болезнь, псориаз и др.
- Болезни, в происхождении которых наследственность не играет роли. Это, например, травмы, ожоги, инфекционные болезни. Генетические факторы в этом случае могут влиять только на течение патологических процессов (скорость регенерации, выздоровления, компенсации функций).
Наследственные заболевания. Геномные, хромосомные и генные мутации
Этиологическими факторами наследственных болезней являются геномные, хромосомные и генные мутации.
Заболевания, обусловленные изменениями структуры молекулы ДНК (генные мутации), называются генными болезнями.
Классификация наследственных болезней
В основу генетической классификации наследственных болезней положен этиологический принцип: тип мутаций и характер взаимодействия со средой. Наследственные болезни подразделяются на 5 групп: генные болезни, хромосомные болезни, болезни с наследственной предрасположенностью (мультифакториальные), генетические болезни соматических клеток и болезни генетической несовместимости матери и плода.
Болезни при несовместимости матери и плода по антигенам возникают в результате иммунологической реакции материнского организма на антигены плода. Наиболее хорошо изученным заболеванием этой группы является гемолитическая болезнь новорожденных, развивающаяся вследствие несовместимости матери и плода по Rh-антигенам. Эта группа составляет около 1% патологии новорожденных.
Особенности клинических проявлений наследственной патологии
Наследственные заболевания часто носят семейный характер. В то же время наличие заболевания только у одного из членов родословной не исключает наследственного характера этой болезни (новая мутация, появление рецессивной гомозиготы).
Для наследственных заболеваний, проявляющихся в любом возрасте, характерно прогрессирующее хроническое течение.
Многие наследственные болезни носят врожденный характер. Не менее 25 % всех форм генных наследственных болезней и практически все хромосомные болезни начинают формироваться уже внутриутробно.
Одним из признаков наследственной патологии является устойчивость к терапии, хотя в некоторых случаях лечение эффективно.
Диагностика наследственных болезней
Диагностика наследственной патологии является сложным и трудоемким процессом и основывается на данных клинического, генеалогического и параклинического обследования.
Наследственные болезни могут протекать сходно с ненаследственными. В некоторых случаях наследственная патология может сопутствовать основному, ненаследственному, заболеванию. Поэтому постановка диагноза включает общее клиническое обследование больного и (при подозрении на конкретную наследственную болезнь) специализированное медико-генетическое обследование.
Однако имеется целый ряд специфических методов, с помощью которых можно изучить вопросы возникновения, развития, распространения, механизмы передачи из поколения в поколение наследственных болезней и роль генотипа и факторов среды в их проявлении.
Клиникогенеалогический метод. Он основан на построении родословных и прослеживании в ряду поколений передачи определенного признака. Этот метод относится к наиболее универсальным методам медицинской генетики.
Близнецовый метод изучения генетики человека позволяет определить роль генотипа и среды в проявлении признаков.
Цитогенетические методы. Основаны на макроскопическом исследовании кариотипа.
Биохимические методы. Основаны на изучении активности ферментных систем либо по активности самого фермента, либо по количеству конечных продуктов реакции, катализируемой данным ферментом. С помощью биохимических нагрузочных тестов можно выявлять гетерозиготных носителей патологических генов, например фенилкетонурии.
Молекулярно-генетические методы. Позволяют анализировать фрагменты ДНК, находить и изолировать отдельные гены и их сегменты и устанавливать в них последовательность нуклеотидов.
Методы гибридизации нуклеиновых кислот. Позволяют выявить специфические фрагменты ДНК, обнаружить единственный ген среди десятков тысяч.
Методы генетики соматических клеток. Позволяют устанавливать группы сцепления у человека, выяснять последовательность расположения генов.
Экспресс-методы. Быстрые предварительные методы изучения генетики ребенка. Часто используются с целью выявления наследственной патологии как скрининг-методы. Например, скрининг новорожденных на фенилкетонурию, гипотиреоз, беременных на альфа-фетопротеин, при помощи которого можно пренатально определить у плода некоторые пороки развития (например, анэнцефалию, открытые формы спинномозговых грыж, синдром Дауна).
Методы пренатальной диагностики наследственных болезней
Пренатальная диагностика связана с решением ряда биологических и этических проблем до рождения ребенка, так как при этом речь идет не об излечении болезни, а о предупреждении рождения ребенка с патологией, не поддающейся лечению (обычно путем прерывания беременности с согласия женщины). При современном уровне развития пренатальной диагностики можно установить диагноз всех хромосомных болезней, большинства врожденных пороков развития, энзимопатий. Часть из них можно установить практически в любом сроке беременности (хромосомные болезни), часть — после 12-й недели (редукционные пороки конечностей, атрезии, анэнцефалию), часть — только во второй половине беременности (пороки сердца, почек).
Показания для пренатальной диагностики: наличие в семье точно установленного наследственного заболевания, возраст матери старше 37 лет, носительство матерью гена Х-сцепленного рецессивного заболевания, гетерозиготность обоих родителей по одной паре аллелей при патологии с аутосомно-рецессивным типом наследования и др.

Наследственные и наследственно-предрасположенные заболевания
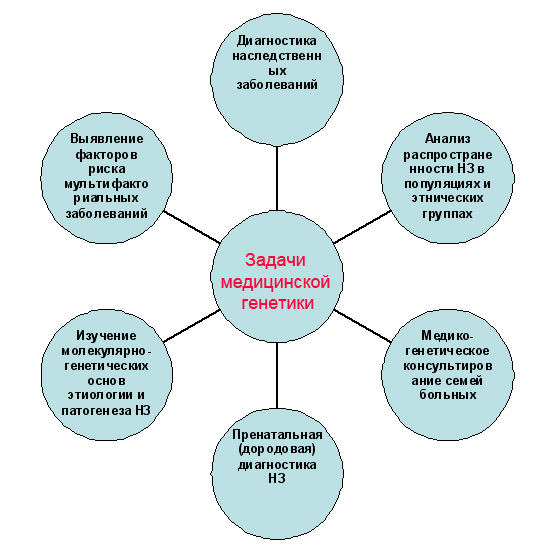
Основной целью медицинской генетики является изучение роли генетических составляющих в этиологии и патогенезе различных заболеваний человека. Эти болезни делятся на два класса: собственно наследственные болезни, куда входят хромосомные и генные заболевания, и болезни с наследственной предрасположенностью, которые называют мультифакториальными заболеваниями.
Хромосомными являются болезни, вызванные нарушением числа, либо структуры хромосом. Генные болезни обусловлены присутствием мутаций в генах. Моногеннными называются болезни, обусловленные присутствием мутаций в одном гене. В этиологии мультифакториальных заболеваний наряду с действием неблагоприятных внешних факторов существенное влияние оказывают состояния не одного, а многих генов. Количество этих генов, формирующих наследственную предрасположенность к заболеванию, иногда исчисляется десятками или даже сотнями. Суммарная частота наследственных заболеваний достигает 1,5%, из них на долю хромосомных болезней приходится 0,5% и на долю моногенных – до 1%. К мультифакториальным относятся большинство наиболее распространенных болезней человека.

Хромосомные болезни: В настоящее время описано около 1000 нозологических форм хромосомных болезней. Все они характеризуются рядом общих признаков, таких как: маленькая масса и длина тела при рождении, пренатальная гипоплазия; отставание в умственном и физическом развитии с момента рождения, особенно выраженное при аутосомных аномалиях; задержка и аномалии полового развития: гипогонадизм, крипторхизм, аменорея, бесплодие и др., более выраженные при аномалиях половых хромосом; множественные ВПР в большей степени при аутосомных аномалиях; комплекс разнообразных по проявлениям и тяжести дизморфогенетических и диспластических признаков, одновременно затрагивающих многие системы и органы больного. Хромосомные болезни редко наследуются, и более чем в 95% случаев риск повторного рождения в семье больного ребенка с хромосомной патологией не превышает общепопуляционного уровня. Исключение составляют те случаи, когда родители больного ребенка несут сбалансированные хромосомные перестройки, чаще всего транслокации, при которых не происходит утраты генетического материала. Носители сбалансированных транслокаций являются практически здоровыми людьми, но вероятность у них выкидышей, замерших беременностей или рождения детей с несбалансированными хромосомными перестройками, а значит с хромосомными болезнями, очень велика. Поэтому при бесплодии, мертворождениях, привычной невынашиваемости беременности, а также при наличии в семье ребенка с хромосомной патологией необходимо проводить анализ кариотипа каждого из родителей с целью диагностики сбалансированных хромосомных перестроек.
Моногенные болезни Разнообразие моногенных заболеваний достаточно велико и их количество по некоторым оценкам достигает 5000. Среди моногенных болезней значительный процент составляют ферментопатии, различные формы умственной отсталости, дефекты органов слуха, зрения, скелетные дисплазии, врожденные пороки развития, болезни нервной, эндокринной, соединительно-тканной, иммунной и других систем. Моногенные варианты течения заболевания в редких случаях встречаются среди любых нозологических форм, которые в общем случае не являются наследственными. Так, например, описаны моногенные формы гипертензии, болезней Альцгейаера и Паркинсона, эпилепсии и других больших психозов, иммунодефицитов, различных онкологических заболеваний и многих других патологических состояний. Моногенные варианты заболевания, как правило, отличаются от спорадических форм более тяжелым течением и ранним дебютом. Большинство мутаций, ассоциированных с моногенными заболеваниями, жестко детерминируют развитие болезни, и факторы окружающей среды не оказывают или оказывают небольшое влияние на развитие заболевания. Поэтому они так трудно поддаются коррекции. Однако немало примеров моногенных болезней с неполной пенетрантностью и варьирующей экспрессивностью, причины которых чаще всего остаются неизвестными. К счастью, моногенные заболевания встречаются достаточно редко. К числу наиболее известных моногенных болезней относятся фенилкетонурия, муковисцидоз, галактоземия, адреногенитальный синдром, гемофилия А и В, миодистрофия Дюшенна/Беккера, проксимальная спинальная мышечная атрофия, гепатолентикулярная дегенерация и многие другие болезни. Профилактика тяжелых неизлечимых моногенных заболеваний проводится на базе пренатальной диагностики.
Мультифакториальные заболевания обусловлены комбинированным действием неблагоприятных внешних и генетических факторов риска, формирующих наследственную предрасположенность к заболеванию. К мультифакториальным заболеваниям относятся подавляющее большинство хронических болезней человека, включая сердечно-сосудистые, эндокринные, иммунные, нервно-психические, онкологические и др. Генетические составляющие могут присутствовать в этиологии даже тех заболеваний, развитие которых целиком индуцируется внешними воздействиями и невозможно без их присутствия, таких, например, как инфекционные болезни. Однако и в этих случаях индивидуальная чувствительность к подобным внешним неблагоприятным воздействиям может быть генетически детерминирована. Например, на сегодняшний день известно, что в патологии бронхиальной астмы, лейкозов и их рецидивов участвуют белковые продукты таких генов системы детоксикации, как GSTM1, GSTT1, CYP1A1, GSTP1, NAT2 и др.Полная расшифровка генома человека открыла большие возможности для изучения ассоциации различных генов человека с моногенными и мультифакториальными заболеваниями. Эти исследования являются основой для планомерной разработки совместно со специалистами различных медицинских профилей новых патогенетических и этиологических методов лечения наследственных заболеваний, а также предупреждения развития тех заболеваний, к которым у человека имеется генетическая склонность.
В настоящее время не существует единой классификации наследственных болезней, и часто их смешивают с врожденными и семейными болезнями. Причиной развития наследственных болезней являются присутствующие в половых клетках родителей мутаций в определенных генах. Эти мутации могут передаваться потомству в ряду поколений. Врожденные заболевания проявляются сразу после рождения, и они могут быть как наследственными, так и приобретенными, например, под действием тератогенных факторов или осложнений в родах. Приобретенные врожденные пороки развития не передаются по наследству. Семейными называются болезни, присутствующие у нескольких членов одной семьи. Они также могут быть наследственными или обусловливаться средовыми влияниями, например неправильным питанием, вредными привычками или присутствием токсических соединений в окружающей среде. В свою очередь, наследственные болезни не обязательно являются врожденными или семейными.
В этиологии детской инвалидности и ограничений жизнедеятельности значительная доля принадлежит наследственным факторам. Так, в Республике Саха (Якутия) среди причин детской инвалидности на первом месте (28,5%) стоят врожденные пороки развития, на втором - заболевания нервной системы (23,9%), на третьем - психические расстройства (11,9%). По данным Росстата среди причин младенческой смертности врожденные пороки развития занимают второе-третье место в Республике Саха (Якутия) и в целом по Российской Федерации. Остается значительной доля врожденных и наследственных заболеваний среди причин детской смертности (в возрасте до 5 лет), в структуре которой на долю хромосомныхболезней приходится 2-3% (Новиков, 2008).
Читайте также: