
Электрофил это в химии кратко
Обновлено: 02.07.2024
В химия, электрофил это химические вещества что образует связи с нуклеофилы приняв электронная пара. [1] Поскольку электрофилы принимают электроны, они Кислоты Льюиса. [2] Большинство электрофилов положительно заряжен, иметь атом, несущий частичный положительный заряд, или атом, у которого нет октета электронов.
Электрофилы в основном взаимодействуют с нуклеофилами через добавление и замена реакции. Часто встречающиеся электрофилы в органический синтез включают катионы Такие как ЧАС + и НЕТ + , поляризованные нейтральные молекулы, такие как HCl, алкилгалогениды, ацилгалогениды, и карбонильные соединения, поляризуемые нейтральные молекулы, такие как Cl2 и Br2, окислители такие как органические перкислоты, химические вещества, которые не удовлетворяют Правило октета Такие как карбены и радикалы, и некоторые кислоты Льюиса, такие как BH3 и ДИБАЛ.
Содержание
Органическая химия
Добавление галогенов
Они возникают между алкенами и электрофилами, часто галогенами, как в реакции присоединения галогена. Общие реакции включают использование бромной воды для титровать по образцу, чтобы определить количество присутствующих двойных связей. Например, этен + бром → 1,2-дибромэтан:
Это принимает форму 3 основных шагов, показанных ниже; [3]
- Формирование π-комплекса Электрофильная молекула Br-Br взаимодействует с богатой электронами молекулой алкена с образованием π-комплекс1.
- Образование трехчленного иона бромония Алкен действует как донор электронов, а бром - как электрофил. Трехчленная ион бромония2 состоит из двух атомов углерода и образует атом брома с выделением Br − .
- Атака бромид-ионом Ион бромония открывается атакой Br − с тыльной стороны. Это дает вицинальный дибромид с антиперипланарный конфигурация. Когда существуют другие нуклеофилы, такие как вода или спирт, они могут атаковать 2 дать спирт или эфир.
Добавление галогенидов водорода
Галогениды водорода, такие как хлористый водород (HCl), добавляют к алкенам с образованием алкилгалогенидов в гидрогалогенирование. Например, реакция HCl с этиленом дает хлорэтан. Реакция протекает с промежуточным катионом, отличным от указанного выше добавления галогена. Пример показан ниже:
Таким образом, стереоселективность продукта, то есть с какой стороны Cl − будет атаковать зависит от типов применяемых алкенов и условий реакции. По крайней мере, какой из двух атомов углерода подвергнется атаке H + обычно решается Правило марковникова. Таким образом, H + атакует атом углерода, который несет меньше заместителей, так что образуется более стабилизированный карбокатион (с более стабилизирующими заместителями).
Это еще один пример ОбъявлениеE2 механизм. [5] Фтористый водород (HF) и йодистый водород (HI) реагируют с алкенами аналогичным образом, и будут получены продукты типа Марковникова. Бромистый водород (HBr) также идет по этому пути, но иногда конкурирует радикальный процесс, и может образовываться смесь изомеров. Хотя во вводных учебниках эта альтернатива упоминается редко, [6] объявлениеE2 обычно конкурирует с ОбъявлениеE3 механизм (более подробно описанный для алкинов ниже), в котором перенос протона и нуклеофильное присоединение происходят согласованным образом. Степень вклада каждого пути зависит от нескольких факторов, таких как природа растворителя (например, полярность), нуклеофильность галогенид-иона, стабильность карбокатиона и стерические эффекты. В качестве кратких примеров, образование стерически необремененного стабилизированного карбокатиона благоприятствует AdE2 путь, в то время как более нуклеофильный ион бромида благоприятствует AdE3 путь в большей степени по сравнению с реакциями с участием иона хлорида. [7]


Гидратация
Один из самых сложных реакции гидратации использует серная кислота как катализатор. Эта реакция протекает аналогично реакции присоединения, но имеет дополнительную стадию, на которой OSO3Группа H заменяется группой OH, образуя спирт:
Как видно, H2ТАК4 принимает участие в общей реакции, однако остается неизменным и классифицируется как катализатор.
Это реакция более подробно:
В целом, этот процесс добавляет молекулу воды к молекуле этена.
Это важная реакция в промышленности, поскольку она производит этиловый спирт, чьи цели включают топливо и исходный материал для других химикатов.
Хиральные производные
Многие электрофилы хиральный и оптически стабильный. Обычно хиральные электрофилы также оптически чисты.
Один такой реагент это фруктоза-производный органокатализатор, используемый в Эпоксидирование Ши. [11] Катализатор может осуществлять высокоэнантиоселективное эпоксидирование транс-дизамещенные и тризамещенные алкены. Катализатор Ши, кетон, окисляется стехиометрическим оксон к активным диоксиран перед тем, как продолжить каталитический цикл.

Оксазиридины например хиральный N-сульфонилоксазиридины влияет на энантиоселективное окисление кетона альфа на пути к сегментам AB-кольца различных натуральные продукты, включая γ-родомиционон и α-цитромицинон. [12]
Связанный полимером хиральный селен электрофилы вызывают асимметричные реакции селененилирования. [13] Реагенты представляют собой арилселененилбромиды, и они были сначала разработаны для химии в фазе раствора, а затем были модифицированы для твердофазного прикрепления шариков через арилокси-фрагмент. Твердофазные реагенты применялись для селененилирования различных алкенов с хорошей энантиоселективностью. Отщеплять продукты от твердой основы можно с помощью оловоорганическое вещество гидридные восстановители. Реагенты на твердой подложке имеют преимущества по сравнению с химией в фазе раствора из-за простоты обработки и очистки.
Шкала электрофильности
| Фтор | 3.86 |
| Хлор | 3.67 |
| Бром | 3.40 |
| Йод | 3.09 |
| Гипохлорит | 2.52 |
| Диоксид серы | 2.01 |
| Сероуглерод | 1.64 |
| Бензол | 1.45 |
| Натрий | 0.88 |
| Некоторые выбранные значения [14] (без размеров) | |
Существует несколько методов ранжирования электрофилов в порядке их реакционной способности. [15] и один из них разработан Роберт Парр [14] с индекс электрофильности ω дано как:
куда р < Displaystyle R ,>это сопротивление (Ом или Ω) и V < Displaystyle V ,>является Напряжение. В этом смысле показатель электрофильности - это своего рода электрофильная сила. Обнаружены корреляции между электрофильностью различных химических соединений и скоростью реакции в биохимических системах и таким явлением, как аллергический контактный дермитит.
Индекс электрофильности также существует для свободные радикалы. [16] Сильно электрофильные радикалы, такие как галогены, реагируют с богатыми электронами реакционными центрами, а сильно нуклеофильные радикалы, такие как 2-гидроксипропил-2-ил и трет-бутил радикальные реагируют с предпочтением бедных электронами реакционных центров.
Суперэлектрофилы
Суперэлектрофилы определяются как катионные электрофильные реагенты со значительно повышенной реакционной способностью в присутствии суперкислоты. Эти соединения впервые были описаны Джордж А. Олах. [17] Суперэлектрофилы образуются как дважды электронодефицитный суперэлектрофил в результате протосольватации катионного электрофила. По наблюдениям Олаха, смесь уксусная кислота и трифторид бора способен удалять гидрид-ион из изобутан в сочетании с плавиковая кислота через формирование суперкислотный из BF3 и ВЧ. Ответственный реактивный промежуточный продукт [CH3CO2ЧАС3] 2+ дикция. Так же, метан может быть нитрован до нитрометан с тетрафторборат нитрония НЕТ +
2 BF −
4 только в присутствии сильной кислоты, такой как фтористоводородная кислота через протонированный дикатион нитрония.
В gitionic (гитон) суперэлектрофилов, заряженные центры разделены не более чем одним атомом, например, ион протонитрония O = N + = O + -H (протонированный ион нитрония). И в дистонический суперэлектрофилы, они разделены 2 и более атомами, например, в реагенте фторирования F-TEDA-BF4. [18]
Понятие кислоты и основания очень близко к понятиям электрофил и нуклеофил, которые широко используются в органической химии для обозначения характера реагирующих частиц.
Нуклеофил – это частица с электронодонорными свойствами, способная образовать связь со своим партнером в реакции (электрофилом), отдавая неподеленую электронную пару или пару связи. Нуклеофилы являются основаниями Льюса.
а) отрицательно заряженные:
Электрофил – это частица с электроноакцепторными свойствами, которая образует связь со своим партнером в реакции (нуклеофилом). У электрофила должна быть вакантная орбиталь или сильнополярная связь. Электрофилы являются кислотами Льюса.
а) положительно заряженные:
Для того чтобы прошла гетеролитическая реакция, нужны два реагента – нуклеофильный (донор электронов) и электрофильный (акцептор электронов).
Понятие кислоты и основания очень близко к понятиям электрофил и нуклеофил, которые широко используются в органической химии для обозначения характера реагирующих частиц.
Нуклеофил – это частица с электронодонорными свойствами, способная образовать связь со своим партнером в реакции (электрофилом), отдавая неподеленую электронную пару или пару связи. Нуклеофилы являются основаниями Льюса.
а) отрицательно заряженные:
Электрофил – это частица с электроноакцепторными свойствами, которая образует связь со своим партнером в реакции (нуклеофилом). У электрофила должна быть вакантная орбиталь или сильнополярная связь. Электрофилы являются кислотами Льюса.
а) положительно заряженные:
Для того чтобы прошла гетеролитическая реакция, нужны два реагента – нуклеофильный (донор электронов) и электрофильный (акцептор электронов).
Электрофильные реакции – реакции органических соединений с электрофильными реагентами, т.е. катионами или молекулами, которые имеют свободную орбиталь, готовые принять электронную пару для образования новой связи.
Примеры электрофильных частиц: H3O + , H + , CH3 + HCl, HNO3, NO2 + , AlCl3, ZnCl2, SO3.
Незаполненность внешнего электронного уровня в электрофиле показана на примере молекулы AlCl3.

Примеры электрофильных реакций:

НУКЛЕОФИ́ЛЬНЫЕ И ЭЛЕКТРОФИ́ ЛЬНЫЕ РЕАГ Е́НТЫ, два типа реагентов в гетеролитических реакциях , различающиеся по характеру их участия в разрыве имеющихся или в образовании новых химич. связей.
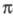
ЭЛЕКТРОФИЛЬНЫЕ РЕАКЦИИ гетеролитич. р-ции орг. соед. с электроф. реагентами (электрофилами, от греч. elektron - электрон и phileo - люблю). К электрофилам относят ионы и молекулы, к-рые имеют достаточно низкую по энергии вакантную орбиталь (льюисовские к-ты) - Н + , D + , Li + , Alk + , AlAlk3, Hal + , BF3, SO3H + , NO + , NO + 2 и др.- и при р-ции с субстратом акцептируют на нее оба связывающих электрона.
В основе Э. р. лежит -электронодонорная способность олефинов, ацетиленов и ароматич. углеводородов по отношению к электрофилам, а также возможность передачи гетероатомами и простыми связями С Ч С и С Ч Н своих электронных пар.
К р-циям электроф. замещения в алифатич. ряду относятся р-ции обмена металлов (гл. обр. ртути) в металлоорг. соед. на другой металл, водород или галоген; водородно-дейтериевый обмен; р-ции изомеризации и др.
Возможны три механизма р-ций: мономолекулярный
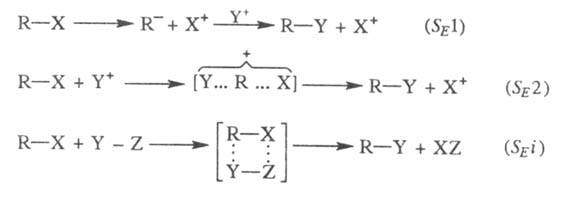
В этих р-циях Y + - электрофил, Х + - электронодефицитная уходящая группа, наз. электрофугом (от лат. fugio -убегаю).
При мономол. процессе в этом случае также наблюдается сохранение конфигурации.
Механизм электроф. замещения зависит от природы, субстрата и р-рителя, как и в случае нуклеоф. замещения. Повышение полярности р-рителя увеличивает возможность ионизации в р-циях, протекающих по механизму влияет гораздо меньше. Большое влияние оказывает природа электрофуга: в случае карбкатионных электрофуюв наблюдается механизм .
Заместители в субстрате, обладающие отрицат. индуктивным и мезомерным эффектами, ускоряют р-ции типа + образует с ароматич. субстратом промежут. комплекс (I). Обычно считают, что для ароматич. соед., активированных электронодонорными заместителями, структура (I) соответствует -комплексу, в к-ром Y* расположен над плоскостью кольца (впервые концепция -комплексов в Э. р. была выдвинута М. Дьюаром в 1946):
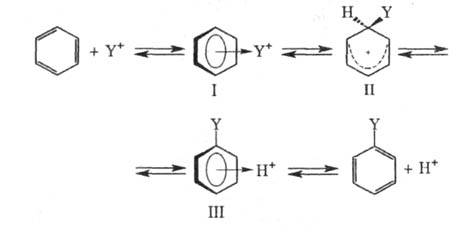
Образование -комплекса, как правило, характеризуется очень высокими скоростями (до 10 10 с -1 ). Лимитирующая стадия - формирование циклогексадиенильного катиона (П), т. наз. -комплекса (комплекса Уэланда, или аренониевого иона), либо, реже, распад II через промежуточный -комплекс III (см. также рис.). Образование -комплексов доказано в нек-рых газофазных р-циях электроф. замещения с помощью радиохим. методов, масс-спектрометрии и ион-циклотронного резонанса; в условиях высокого вакуума -комплексы м. б. достаточно устойчивыми. Из р-ров препаративно выделены соли ряда катионов II. При использовании высокореакционноспособных реагентов лимитирующей стадией может стать стадия образования -комплекса.
В р-циях электроф. замещения монозамещенных производных бензола новая группа вступает в орто-, мета- или пара -положение; при этом заместители либо облегчают, либо затрудняют протекание р-ции. По правилам ориентации заместители, проявляющие положит. индуктивный эффект (+I) и положит. мезомерный эффект (+М), активируют ароматич. ядро и являются орто- или пара -ориентантами. Заместители, проявляющие -I и +М-эффекты, также орто- или пара -ориентанты; однако, в случае когда индуктивный эффект больше мезомерного (напр., у Hal), ароматич. ядро пассивируется, если же индуктивный эффект меньше мезомерного (OR, SR, NR2 и др.), - ядро активируется. Заместители, проявляющие -I и -М-эффекты (CN, NO2), пассивируют ядро и являются мета -ориентантами (см. табл.). По иной классификации орто- и пара -ориентанты относят к заместителям I рода, а мета -ориентанты - к заместителям II рода. Причина разл. реакц. способности орто-, мета- и пара -положений - изменение распределения электронной плотности в кольце под влиянием заместителей.
Читайте также: