
Роль протеомики в оценке патологических состояний кратко
Обновлено: 17.06.2024
Протеомика – это медико-биологическая наука, занимающаяся изучением совокупности всех белков клетки (органа, ткани, организма), их взаимодействия между собой и реакций на изменяющиеся условия внешней или внутренней среды.
Технологии протеомики в конце 20 века позволили достичь большого прогресса в научных исследованиях различных отраслей биологии и медицины, результаты которых затем стали успешно применяться в медицинской практике.
На современном этапе для крупномасштабных исследований белков обычно применяются протеомные технологии, основанные на масс-спектрометрии, и метод двумерного электрофореза.
Двумерный электрофорез является методом наиболее распространенным, имеющим высокое разрешение, позволяющим параллельно отделять белки от различных смесей. Также этот метод сопровождается выборкой и окрашиванием дифференцированных белков, определяемых масс-спектрометрией. Несмотря на широкое применение у этой технологии есть и свои недостатки, например, неспособность отделить все белки образца ввиду их различий в уровне экспрессии, результатом чего становится отличие их свойств.
Главные задачи протеомных технологий:
- Изучение всех белков, содержащихся в исследуемом образце (их количественные характеристики и качественное определение (структуры)). Некоторые технологии могут определить только количественную составляющую.
- Проведение аналитического фракционирования полипептидных цепей по двум независимым друг от друга физико-химическим свойствам, которые отражают особенности их первичной структуры.
- Выявление различных белковых фракций путем их разделения (используются высокочувствительные методы детекции белков – некоторые модификации масс-спектрометрии).
- Систематизация полученных данных путем стандартизированного описания белков.
Значение технологий протеомики для практической медицины
Практическое значение протеомики для медицины:
Множество идей применения протеомики в медицинской сфере находится только на стадии разработки. Например, предлагается ведение протеомных карт биологических жидкостей, в первую очередь крови. При наличии изменений белков крови в динамике можно будет заподозрить и выявить заболевание и своевременно начать лечение.
Первичная структура белка — последовательность расположения аминокислотных остатков в полипептидной цепи, составляющей молекулу белка. Связь между аминокислотами — пептидная.Если молекула белка состоит всего из 10 аминокислотных остатков, то число теоретически возможных вариантов белковых молекул, отличающихся порядком чередования аминокислот, — 1020. Имея 20 аминокислот, можно составить из них еще большее количество разнообразных комбинаций. В организме человека обнаружено порядка десяти тысяч различных белков, которые отличаются как друг от друга, так и от белков других организмов.Именно первичная структура белковой молекулы определяет свойства молекул белка и ее пространственную конфигурацию. Замена всего лишь одной аминокислоты на другую в полипептидной цепочке приводит к изменению свойств и функций белка. Например, замена в β-субъединице гемоглобина шестой глутаминовой аминокислоты на валин приводит к тому, что молекула гемоглобина в целом не может выполнять свою основную функцию — транспорт кислорода; в таких случаях у человека развивается заболевание — серповидноклеточная анемия.
При различных заболеваниях происходит изменение белкового состава тканей. Эти изменения называются протеинопатиями. Различают наследственные и приобретённые протеинопатии.Наследственные протеинопатии развиваются в результате повреждений в генетическом аппарате данного индивидуума. Какой-либо белок не синтезируется вовсе или синтезируется, но его первичная структура изменена. Примеры наследственных протеинопатии - гемоглобинопатии
6. Роль протеомики в оценке патологических состояний
Протеомика в повседневной жизни
Однако, несмотря на значительность проекта, данные о геноме человека и других организмов поставили перед учеными новые, более глобальные задачи. Оказалось, что генов в организме человека в несколько раз меньше, чем ожидалось.
Их количество составляет порядка 35 тыс., в то время как число кодируемых в геноме белков – порядка миллиона. Многообразие белков обусловлено наличием таких сложнорегулируемых процессов, как процессинг мРНК, посттрансляционные модификации и процессинг белков. Невозможность получения полной информации о составе белков организма с помощью геномики явилось основной предпосылкой развития постгеномных исследований и возникновения новой науки – п
ротеомики, изучающей состав, структуру и функции экспрессированных геномом белков и белковых комплексов. Клетка реагирует на изменения внешней среды изменением протеома (набора белков): в ответ на воздействие синтез одних белков увеличивается, а других уменьшается. Следовательно, протеом отражает информацию о состоянии организма при определенных физиологических условиях и в определенный момент времени. Именно протеом обусловливает в итоге функцию каждой клетки.
Белковая патология является причиной 98,5% известных заболеваний, 95% всех фармакологических средств адресовано белкам. Следовательно, особый интерес представляют медицинские аспекты реализации протеомных исследований. Известно, что эффективность лечения многих заболеваний (онкологических, сердечно-сосудистых, бронхолегочных, нейродегенеративных, эндокринных и др.) зависит от своевременной и точной диагностики. Поэтому усилия ученых направлены на поиск белков – маркеров заболеваний, имеющих диагностическое и терапевтическое значение, и на разработку новых эффективных диагностических методов и лекарственных средств.
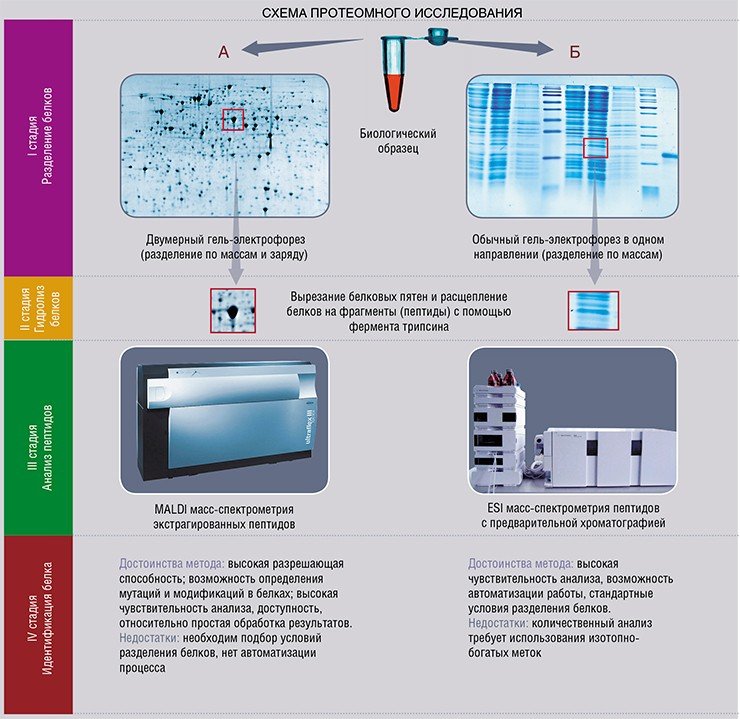
Основным инструментом протеомики является метод двумерного гель-электрофореза в сочетании с масс-спектрометрией, который позволяет проанализировать несколько тысяч белков в одном образце и детектировать изменения их концентраций. Полученная таким способом протеомная карта образца отражает белковый состав клетки, ткани или даже целого органа. Для обнаружения потенциальных белков-маркеров заболеваний используется сравнительный анализ протеома биологических жидкостей, клеток, тканей и органов человека (рис. 1).
Особый интерес для клинической диагностики вызывает разработка протеомных биочипов, позволяющих проводить широкомасштабные исследования биологических образцов на наличие биомаркеров заболеваний. Протеомный биочип представляет собой матрицу, к которой химически присоединены молекулы, способные специфически взаимодействовать только с искомым белком-маркером с полным удалением из образца всех остальных веществ. Детекцию связавшегося белка производят высокоточным масс-спектрометрическим методом. Такой подход увеличивает производительность диагностических методов, снижает их себестоимость, позволяет анализировать одну пробу на наличие нескольких биомаркеров за короткое время. В Институте биофизики и клеточной инженерии совместно с сотрудниками Института физико-органической химии НАН Беларуси проводятся работы по созданию биочипов для раннего обнаружения и мониторинга сахарного диабета с использованием масс-спектрометрической детекции.
По словам нобелевского лауреата в области физиологии и медицины Ли Хартвела, именно протеомная диагностика является диагностикой будущего, ведущей к персонифицированной медицине. Выбор схем терапии и подбор лекарственных схем часто проводится вслепую, методом проб и ошибок. Так как именно белок является мишенью, на которую направлено действие лекарственных средств, то биомаркеры можно использовать не только в диагностических целях, но и для поиска и конструирования медицинских терапевтических препаратов. Число потенциальных белков-мишеней действия лекарств исчисляется тысячами, тогда как коммерчески используется лишь несколько сотен. Обнаружение новых мишеней приведет к созданию более эффективных и безопасных медикаментов. Сегодня в составе практически всех крупных фармацевтических компаний (Pfizer, Roche, Siena Biotech, Merck и др.) имеются подразделения, занимающиеся протеомными исследованиями. На создание каждого лекарства традиционными методами необходимо более 10 лет работы и сотни миллионов долларов. При этом 90% лекарств-кандидатов не попадают на рынок, отсеиваясь в процессе разработки. Несмотря на строгий отбор и контроль качества медикаментов, в Европе около 15% случаев госпитализации обусловлено их побочным действием. Протеомика позволяет ускорить процесс разработки лекарственных средств, обеспечивая не только идентификацию мишеней, но и изучение метаболизма препаратов, тестирование их эффективности, токсичности и индивидуальной восприимчивости. Очевидно, что интеграция персонифицированной медицины, целью которой является индивидуальный подход к терапии каждого конкретного пациента, а не лечение болезни в целом, и протеомики приведет к революции в здравоохранении и фарминдустрии. Ведь такой системный подход обеспечит индивидуализацию фармакотерапии, что повысит качество, результативность и безопасность лечения.
В Институте биофизики и клеточной инженерии ведется работа по созданию клеточной системы для мониторинга биологического действия техногенных наноматериалов на организм человека с применением протеомных методов. Поскольку нанотехнологии все чаще применяются в медицине, пищевой и легкой промышленности, это обусловливает актуальность разработки тест-систем для детального анализа механизмов взаимодействия наноматериалов с живыми организмами.
Протеомные методы применяются в исследованиях, касающихся пищевой промышленности и сельского хозяйства. Это выявление маркеров, отвечающих за качество сельскохозяйственной продукции (питательные и вкусовые характеристики мяса, молока), идентификация диагностических белков, ответственных за устойчивость растений и животных к патогенам, диагностика заболеваний сельскохозяйственных животных на ранних стадиях (мастит, заболевания желудочно-кишечного тракта). Протеомные методы апробируют для контроля свежести продуктов и определения присутствия в них патогенных микроорганизмов, характеристики сортов вина, пива, сыра и детекции качества сырья, из которого они приготовлены, выявления аллергенов во фруктах, овощах, продуктах питания.
Протеомные исследования актуальны для процесса селекции в сельском хозяйстве, включая идентификацию и отбор определенных пород животных и сортов растений. Так, с использованием методов сравнительной протеомики нами идентифицированы диагностические маркеры, характеризующие устойчивость сортов томата к фитофторозу. Совместно с НПЦ НАН Беларуси по картофелеводству и плодоовощеводству продемонстрирована эффективность протеомного подхода для сортовой идентификации соматических гибридов картофеля. Сейчас в институте проводятся эксперименты по идентификации белков, отвечающих за мясную продуктивность и хозяйственно полезные характеристики мяса, а также за развитие стрессоустойчивости сельскохозяйственных животных.
Неоценима помощь протеомики и в фундаментальных исследованиях, в частности при анализе процессов внутриклеточной сигнализации в животных и растительных клетках. В Институте биофизики и клеточной инженерии из клеток растений впервые выделены и масс-спектрометрически идентифицированы белки-мишени действия циклического гуанозинмонофосфата, являющегося универсальным вторичным медиатором. Результаты найдут применение при разработке методов оптимизации процессов выращивания сельскохозяйственных растений, а также для создания генно-модифицированных растений с улучшенными характеристиками.
В лаборатории молекулярной биологии клетки получен трансгенный картофель со стабильным наследованием и экспрессией целевых синтетических генов, отвечающих за синтез антимикробных пептидов, и работы по созданию генно-модифицированных растений с заданными свойствами продолжаются. Актуальными представляются исследования по оценке их биобезопасности, которые планируется осуществить путем анализа протеомного статуса с целью выявления возможных изменений белкового состава под действием встроенных и модифицированных генов. Данные изыскания лягут в основу разработки протеомного диагностикума хозяйственно полезных признаков животных и растений для эффективного селекционного отбора.
Протеомика активно развивается во всем мире и, несмотря на свою относительную молодость, достигла впечатляющих успехов. Сделан значительный прогресс в области протеомной диагностики ряда заболеваний, включая наиболее распространенные формы рака (рак яичника, желудка, простаты и т.д.), заболевания сердечно-сосудистой системы (инфаркт миокарда, ишемическая болезнь сердца), нейродегенеративные заболевания (шизофрения, болезнь Альцгеймера и т.д.). Разработаны и используются тесты для диагностики рака мочевого пузыря (NMP22 bladder cancer test, Matritech Inc.), оценки эффективности химиотерапии при раке молочной железы (DirectHit Panel for Breast Cancer, CCC Diagnostics Inc.).
Многообещающие перспективы протеомика открывает и в области фарминдустрии. C использованием методов компьютерной протеомики были разработаны лекарственные препараты для терапии СПИДа (Saquinavir, Ritonavir, Indinavir, Nelfinavir, Amprenavir, Fosamprenavir), глаукомы (Trusopt), рака (Thymitaq, Prinomastat, Gleevec), гриппа (Tamiflu, Zanamivir) и др. Протеомное профилирование спектра мишеней, с которыми взаимодействует Gleevec, изначально разработанный компанией Novartis как высокоизбирательный препарат для лечения хронической миелоидной лейкемии, выявило новые мишени действия данного лекарства, что позволило применять Gleevec и для терапии опухолей желудочно-кишечного тракта.
Однако внедрение полученных результатов в повседневную медицинскую практику для диагностических и прогностических целей, подбора индивидуальных схем терапии возможно только при тесной кооперации специалистов разных областей – ученых, медиков, программистов, производителей лекарств и др. – с привлечением как государственных организаций, так и коммерческих структур для всесторонней оценки и интерпретации результатов. Так, в США ратификацией лекарственных средств, в том числе разработанных с помощью протеомных технологий, занимается специальная правительственная структура – Управление по контролю за качеством пищевых продуктов и лекарственных препаратов (Food and Drug Administration, FDA).
Несомненно, есть ряд ограничений, лимитирующих скорость развития протеомики. Например, по сравнению с геномикой сегодня не существует аналога полимеразной цепной реакции для белковых молекул, что обусловливает лимит детекции в протеомике, составляющий 10-2 М. Еще одной проблемой является необходимость определения белков в биологическом образце, в котором в высокой концентрации присутствуют другие белки (например, разница в концентрациях между белками плазмы крови может достигать 1010–1012). Не вызывает сомнения и тот факт, что для высокопродуктивного скрининга белковых молекул необходимы приборы не только с большей чувствительностью, но и с большей производительностью.
Чрезвычайная многоаспектность реализации протеомных исследований в фундаментальных и прикладных областях еще раз указывает на масштабность задач, стоящих перед протеомикой завтрашнего дня. Непрерывное совершенствование протеомных методов, повышение чувствительности аналитического оборудования, автоматизация исследований, несомненно, приведет к полному пониманию молекулярных механизмов функционирования белковых систем, что позволит в будущем целенаправленно управлять этими процессами для оценки статуса организма и коррекции патологических состояний. Протеомика открывает неограниченные возможности для медицины, сельского хозяйства, ветеринарии, пищевой промышленности, фарминдустрии и других смежных областей.

Термин "точная медицина" относится к использованию диагностических, терапевтических и мониторинговых стратегий для отдельных пациентов на основе их молекулярных профилей. В медицине (на практике) фокус исследований сосредоточен главным образом на стратификации болезней на подтипы, основанные на молекулярных биомаркерах или сигнатур, т.е. в молекулярной таксономии болезни. Цель современного врача состоит в том, чтобы использовать эти сигнатуры для назначения правильного лечения пациентам, распределения их в определенные подгруппы заболеваний и назначения наиболее эффективной терапии для каждого пациента. Обширная молекулярная характеристика признаков экспрессии генов при раке молочной железы позволила разработать мультигенные анализы, которые в настоящее время проходят клинические испытания для рутинного использования в клинике с целью руководства лечением пациентов и мониторинга.
Хорошо известно, что изменения в экспрессии генов не всегда отражают изменения в содержании белка. Белки являются основными факторами, влияющими на функции клеток, благодаря изменениям их посттрансляционных модификаций и их распространенности, что отражается также на изменениях их интерактома с влиянием на фенотипы клеток. Поэтому очень важно также учитывать протеомику, фосфопротеомику и другие наборы данных PTM-'omics 'в исследованиях для того , чтобы понять развитие и подтипы заболевания, поскольку они могут лучше отражать функциональное состояние и динамические свойства клетки.
Последние достижения в области протеомики позволяют точно измерять содержание тысяч белков и фосфопротеинов из нескольких образцов параллельно. Таким образом, впервые у нас появилась возможность систематически измерять протеомные профили тысяч образцов пациентов или клеточных линий модели заболевания, выявлять точный лежащий в их основе молекулярный механизм, открывать персонализированные биомаркеры, сети и методы лечения. В этой заметкерассматриваются примеры успешного использования наборов данных протеомики и фосфопротеомики, а также их интеграции с другими наборами данных различных омик для точной медицины. Здесь также обусждаются проблемы биоинформатики, анализ и интеграция таких больших наборов данных и потенциальные причины, по которым профилирование протеомики и биомаркеры в настоящее время широко не используются в клинических условиях. Однако эти наборы данных не были использованы в области точной медицины из-за времени, необходимого для отбора, сложности и динамического диапазона образцов протеомики, отсутствия воспроизводимости среди лабораторий, различий между методами количественного определения и другими смешанными факторами.
В последнее время технологические разработки в области приборостроения, подготовки проб и анализа данных и инициативы по разработке стандартов для сбора и оценки этих данных привели к появлению высококачественной, воспроизводимой и всеобъемлющей протеомики и созданию протоколов для генерации таких данных. Исследователи смогли обнаружить 50 000 фосфопептидов в одной линии раковых клеток человека, и теперь ученые могут регулярно и точно измерять тысячи пептидов в короткие сроки. Hebert et al. ( 2014) смогли измерить весь протеом дрожжей, содержащий пептиды из белков 803980, всего за 1 час. Сотни целевых и глобальных наборов данных протеомики также собираются CPTAC (Консорциум клинического анализа протеомных опухолей) для того , чтобы внести свой вклад в изучение рака. Таким образом, сообщество биоинформатиков должно в настоящее время решить проблему использования этого нового слоя информации( протеомики ) и интеграции его с другими ценными слоями омики для изучения механизма развития заболеваний человека.
Целевые методы протеомики, такие как SRM / MRM (мониторинг выбранных / множественных реакций) и независимые от данных методы сбора , такие как SWATH-MS (последовательное оконное получение всех теоретических фрагментов масс-спектров ионов), также позволяют значительно понизить уровень изменчивости во время сбора данных и улучшить качество набора данных.
Основное применение протеомики - идентификация биомаркеров заболевания. В идеале биомаркер должен однозначно распознавать заболевание и должен обнаруживаться в доступной жидкости организма, такой как плазма, кровь, сыворотка мочи, слюна или спинномозговая жидкость. Например, простат-специфический антиген (ПСА, PSA) является одним из самых известных неинвазивных скрининговых биомаркеров и используется для выявления рака предстательной железы. Однако, высокая концентрация ПСА в крови также связана с доброкачественной гиперплазией предстательной железы и простатитом. Таким образом, даже несмотря на то, что PSA обеспечивает достаточную чувствительность, он не способен различать рак простаты и другие патологии простаты из-за его низкой специфичности. В последние годы, чтобы улучшить чувствительность и специфичность биомаркеров, исследователи обратились к комбинации биомаркеров, то есть сигнатуры заболевания, вместо того, чтобы искать идеальный биомаркер.
Используя протеомическую характеристику образцов из разных стадий прогрессирования рака молочной железы люминального типа, исследователи выявили различия в компонентах белкового гомеостаза и метаболической регуляции, которые способны отличать здоровые, от первичных или метастазирующих в лимфатических узлах опухолевых тканей, а также от лимфатических узлов и рака молочной железы. Основанное на протеомике субтипирование пациентов с раком толстой кишки и прямой кишки с помощью CPTAC также оказалось более подробном , чем на основе данных транскриптомики, что привело к лучшему прогнозированию течения онкологического заболевания. Комбинация белка с измерениями содержания фосфопротеинов с использованием белковых массивов с обращенной фазой также использовалась, например, для прогнозирования рецидива рака яичников. Многочисленные исследования показали ценность данных фосфопротеомики для информации, лежащей в основе механизма болезни. Например, данные фосфопротеомики были использованы для выявления механизма устойчивости клеток меланомы к ингибиторам BRAF и глиобластомы к ингибиторам mTOR ( мишень рапамицина), что привело к открытию новой комбинированной терапии. Клеточно-специфическая фосфопротеомика также была использована для изучения двунаправленной передачи сигналов между эндотелиальными клетками и опухолевыми клетками, чтобы понять метастатические механизмы опухолевых клеток. Поэтому ясно, что протеомный и фосфопротеомический слой информации о омиках может дать ценную информацию в нашем стремлении к точной медицине.
Один из подходов к выявлению биомаркеров заключается в использовании тех белков, которые по-разному экспрессируются между нормальным и болезненным состоянием. Также используются более сложные методы, такие как машинное обучение и сетевые подходы. Методы машинного обучения, такие как метод опорных векторов , нейронные сети , "дерево решений" , "случайный лес" и генетические алгоритмыбыли успешно применены к данным протеомики для идентификации биомаркеров нескольких типов рака, сердечной недостаточности и других патологических состояний. Кроме того , исследователи сконструировали 29-плексную матричную платформу, включающую 29 потенциальных биомаркеров, связанных с аденокарциномой желудка.
Наборы протеомных данных с высокой пропускной способностью характеризуются большим количеством переменных / характеристик по сравнению с общим количеством доступных образцов. Следовательно, входное пространство включает в себя множество не относящихся к делу или шумных элементов, которые в сочетании с широкой неоднородностью, обычно встречающейся в биологических образцах, затрудняют идентификацию действительно важных биомаркеров. Для решения этой проблемы использовались методы уменьшения размерности , такие как PAM (анализ прогнозирования для микрочипов), SVM-RFE (устранение машинно-рекурсивных характеристик опорных векторов) , SAM (анализ значимости микрочипов) , которые используются в сочетании с методами машинного обучения для уменьшения шума в наборах данных. Это достигается за счет отказа от несущественных признаков и повышения эффективности обобщения и прогнозирования.
Отсутствие воспроизводимости в различных наборах данных, технические проблемы, такие как проблема переоснащения в подходах машинного обучения и внутренняя сложность заболеваний человека, часто не позволяют перспективным биомаркерам достичь клинического применения.
Раскрытие индивидуальных механизмов развития и прогрессирования заболевания у разных пациентов может стать ключом к разработке точных стратегий терапии. В качестве первого шага в этом направлении подходы омического анализа данных (например, гены, белки или фосфопептиды) обычно пытаются идентифицировать затронутые биологические процессы и функции , используя генную онтологию или анализ обогащения пути (или других признаков) на дифференциально регулируемых объектах каждого из них. Эти дифференциально регулируемые объекты также могут быть отображены на существующие сети взаимодействия или карты маршрутов, чтобы обеспечить лучшее представление о процессах происходящих в клетках. Например, в исследовании двунаправленной передачи сигналов опухоли эндотелия, авторы нанесли пораженные фосфопептиды на карты путей KEGG , чтобы понять пути, вовлеченные в трансэндотелиальный метастаз опухолей.
Совсем недавно была собрана коллекция методов, в основном разработанных и применяемых к наборам данных геномики и транскриптомики, которые учитывают также сеть взаимодействия белков и структуру биохимических путей для выявления специфических для пациента звеньев патогенеза болзени. Алгоритм SPIA (Анализ воздействия сигнального пути) объединяет информацию о дифференциальной экспрессии генов с их влиянием в этом пути на основе их размещения в его топологии. HotNet2 и связанная диффузия через взаимодействующие события (TieDIE) помогла использовать слегка измененные диффузионные подходы, которые включают в себя форму случайного блуждания и взвешивания в соответствии с силой соединения и топологией сети для распространения эффекта возмущения в данной сети.
Несмотря на обширную информацию, которую могут предоставить данные протеомики и фосфопротеомики, она все еще представляет собой только один слой функций и регуляции клеток. Таким образом, чтобы действительно глубоко понять функцию клетки, важно рассмотреть как можно больше уровней регуляции функции клетки. Это особенно верно в контексте точной медицины, где разные уровни клеточной регуляции могут быть важны для каждого пациента. В настоящее время и следует это признать не существует стандартного или оптимального подхода к интеграции данных. В зависимости от наборов данных, которые они интегрируют, методы можно разделить на однородные, где наборы данных содержат данные одного типа, но из разных источников, и разнородные, где интегрированы несколько наборов данных с различными типами данных. Эти методы могут интегрировать слои информации поэтапно или шаг за шагом для создания интегрированной модели исследуемой системы. Например, Drake et al. (2014) использовали пошаговый подход для интеграции геномных, транскриптомных и фосфопротеомических данных для того , чтобы идентифицировать специфические для пациента сети, которые поражены раком предстательной железы, и предложить потенциально точный метод лечения для этих пациентов. В частности, они сначала использовали наборы данных, чтобы идентифицировать пути, факторы транскрипции и киназы, которые, вероятно, активны в своих образцах, а затем применили их алгоритм на основе диффузии, TieDie , чтобы определить различные функциональные модули и пути, которые затрагиваются у разных пациентов. В этом исследовании они также показали, что интеграция фосфопротеомики была в состоянии обнаружить пути, которые в противном случае были бы упущены, подчеркивая важность включения этого уровня информации в подходы точной медицины. Применяя этот конвейер на трех различных клеточных линиях простаты в качестве валидации, они смогли подтвердить свои результаты либо путем оценки их прогнозируемой чувствительности к лекарственным средствам, либо с помощью исследований сущности генов.
Белковое изобилие
В отличие от геномов, протеомы, т. е. полные наборы белков клетки, представлены активным набором молекул, которые постоянно модифицируются. При этом, если биохимия имеет дело с отдельными выделенными молекулами, то в случае с протеомом мы имеем дело с огромным молекулярным пулом (уместно провести аналогию с рыбой, пойманной на удочку, и рыбным изобилием принесенным неводом).
Функциональная, структурная и медицинская
Протеомика является классическим образцом междисциплинарной науки: она объединяет биологию, химию, компьютерное моделирование, сложную инструментальную технику
На официальной странице HUPO подробно изложены основные направления исследований, перечень которых может многое сказать даже неспециалисту: протеом человека, протеомика мозга, изучение антител, болезни, вызванные нарушениями метаболизма сахаров, протеомика сердечно-сосудистых заболеваний, протеомика стволовых клеток, определение биомаркеров заболеваний, изучение заболеваний человека на мышиных моделях и т. д.
Методологически в протеомике выделяют несколько направлений, главными среди которых являются функциональная, структурная и медицинская (клиническая) протеомика.
О целях функциональной протеомики уже упоминалось выше. Это получение информации о межбелковых взаимодействиях и их влиянии на экспрессию и модуляцию активности генов, а также пост-трансляционную модификацию белков в составе белковых комплексов.
Структурная протеомика, несмотря на то что является классическим направлением исследования белков, тем не менее, продолжает активно развиваться вследствие усовершенствования аналитических методов, таких как новые варианты ЯМР-спектроскопии, рентгеноструктурного анализа и масс-спектрометрии.
Современные достижения
Над поиском протеомных маркеров значимых заболеваний интенсивно работают исследователи всего мира – не только ученые из академических институтов, но и специалисты из исследовательских подразделений крупных и средних фармацевтических компаний.
Уже достигнуты определенные успехи в одной из самых проблемных областей медицины – ранней диагностике тяжелых заболеваний. В первую очередь это относится к раку предстательной железы (Downes et al., 2007; Larkin et al., 2010). Диагностика этого широко распространенного заболевания, проводящаяся по наличию в моче пациента белка простатспецифического антигена, – на сегодняшний день одна из самых ранних и точных.
К настоящему времени достигнуты неплохие результаты и в выявлении маркеров рака молочной железы (Mathelin et al., 2006; Gast et al., 2009). Из-за широкой клинической вариабельности этого заболевания в качестве маркеров предлагается использовать набор из 40 белков. Такой белковый профиль позволяет не только с высокой точностью диагностировать заболевание, но и прогнозировать эффективность лечения. Основные диагностические белки этого набора – гаптоглобин, трансферрин, аполипопротеины A-I и C-I – уже сегодня используются при диагностике рака молочной железы.

Ведутся исследования и по обнаружению маркеров нейродегенеративных заболеваний, таких как болезнь Альцгеймера, склерозов различной этиологии и т. д. (Cedazo-Minguez, Winblad, 2010). В этой области основными прогностическими маркерами являются ангиогенин (фермент, обеспечивающий рост кровеносных сосудов), креатининкиназа, фибриноген, аполипопротеин Е (Bowser, Lacomis, 2009)
В Сибири проблемами структурной и функциональной протеомики интенсивно занимаются в новосибирском Институте химической биологии и фундаментальной медицины СО РАН. В результате совместной работы с НИИ Психического здоровья ТНЦ СО РАМН проведена большая работа по поиску белков-маркеров шизофрении.
Этиология и патогенез этой тяжелейшей психической болезни неизвестны. Согласно одной из теорий возникновения шизофрении, в основе заболевания лежит нарушение белкового обмена. Сравнение протеомных профилей статистически достоверной выборки людей, страдающих шизофренией, и протеомных профилей здоровых добровольцев уже позволило исследователям выявить опредленный набор белков в качестве маркеров: аполипопротеин A-II, фосфомевалонат киназу и сериновую (треониновую) киназу. Дальнейшие усилия ученых будут направлены на уточнение роли этих белков в патогенезе болезни и внедрение этих маркеров в клиническую биохимию.
Cox J., Mann M. Is proteomics the new genomics? // Cell. 2007. V. 130(3). P. 395—398.
Capelo J.L., Carreira R., Diniz M. et al. Overview on modern approaches to speed up protein identification workflows relying on enzymatic cleavage and mass spectrometry-based techniques // Anal. Chim. Acta. 2009. V. 650. N 2. P. 151—159.

В Протеомика это наука , которая изучает протеома , то есть все белки в клетке, в органелл , ткани, органа или организма в какой - то момент и при данных условиях.
На практике протеомика пытается идентифицировать глобальным образом белки, извлеченные из клеточной культуры, из ткани или из биологической жидкости, их расположение в клеточных компартментах, их возможные посттрансляционные модификации, а также их количество.
Это позволяет количественно оценить вариации в уровне их выражения в зависимости от времени, окружающей среды, состояния развития, физиологического и патологического состояния , вида происхождения. Он также изучает взаимодействия белков с другими белками, с ДНК, РНК или другими веществами.
Функциональная протеомика изучает функции каждого белка.
Протеомика также исследует структуру первичного , вторичного и третичного белка.
Резюме
История
Были разработаны многие методы, которые до сих пор широко используются.
Техника электрофореза была разработана в 1892 г. С. Е. Линдером и Х. Пиктоном. Принцип хроматографии восходит к 1861 году Фридрихом Гоппельсредером .
// поставить фреску с хронологией изучения белков
За последние десять лет протеомика стала самостоятельной наукой со своими собственными методами и методами. Протеомика была награждена в 2002 году Нобелевской премией по химии.
Она позаимствовала многие технологии, ранее использовавшиеся в других дисциплинах, и применила их для изучения белков. Можно упомянуть, например, об использовании масс-спектрометрии , основанной на физическом и химическом анализе , при идентификации белков , при количественной оценке экспрессии белков , при локализации пептидов в ткани, в поисках конкретных биомаркеры из патологий .
Почему протеом?
- Большие белки часто имеют сложную пространственную структуру, где компактные субъединицы связаны вместе гибкими цепями, которые играют роль во взаимодействиях между белками или другими веществами. Рентгеноструктурный анализ белков показывает, что они построены вокруг жесткой кристаллической решетки, которая предотвращает или снижает гибкость субъединиц.
- Сумма информации, полученной в результате геномного анализа, не отвечает на все вопросы, связанные с протеомным анализом (сложным и дорогим).
Ключевые события будут вести от запаса информации, составляющей геном, к производству молекул, которые будут определять и регулировать жизнь клеток, белков. Теоретически последовательность каждого гена будет транскрибироваться (или нет) в информационную РНК (мРНК), которая сама транслируется в белок. Фактически гены в эукариотических клетках часто фрагментированы и содержат участки ( интроны ), отсутствующие в мРНК. Частичная транскрипция будет разрешена путем сплайсинга РНК-предшественницы, копии гена, который может дать начало различным мРНК, каждая из которых может привести к нескольким белкам. Таким образом, мы можем выдвинуть ряд доводов в пользу развития протеомного анализа:
- Идентификация и оценка уровней белка имеют решающее значение для получения полной картины многих биологических процессов.
- Однако изобилие белков внутри клетки регулируется не только на уровне транскрипции, но также на трансляционном и посттрансляционном уровнях, так что невозможно установить простую взаимосвязь между уровнем d 'MRNA и белком. Тот факт, что один ген и даже одна мРНК может привести к нескольким белкам, различающимся по функциям, делает эту корреляцию опасной.
- Наконец, некоторые белки имеют длительный срок жизни, то есть даже синтезированные с низкой скоростью они могут накапливаться в клетке, оставаясь при этом функциональными, в то время как другие - с коротким сроком жизни - быстро удаляются. Так что, даже если их синтез будет быстрым, а в конечном итоге они будут медленными.
- Белок в соответствии с его клеточным состоянием (дифференциация, пролиферация, апоптоз ) будет обнаружен в данном компартменте ( цитоплазме , ядре, митохондриях ) или будет секретироваться клеткой. Без анализа протеома изменение локализации белка, необходимое для его биологической активности, останется незамеченным.
- Большинство белков становятся биологически активными только после стадий ко- и посттрансляционного созревания (таких как гликозилирование , фосфорилирование , дезаминирование и т. Д.). Они также являются индикаторами состояния клеточного аппарата.
Различные подходы к протеомике
На практике белки сначала извлекаются из популяции клеток или ткани, а затем отделяются перед идентификацией.
Добыча
Первым шагом обычно является извлечение белков из биологического образца. Этот шаг имеет решающее значение: неправильная экстракция может привести к деградации белка и затруднить или даже сделать невозможным идентификацию белков. Эти мембранные белки , имеющие множество аминокислот гидрофобными и , следовательно , плохо растворим, с трудом поддаются изучению.
Некоторые методы не требуют извлечения белков из исследуемой ткани. При иммунолокации ткань фиксируется, а затем разрезается на тонкие полоски толщиной в несколько микрон. Затем белки обнаруживаются in situ мечеными антителами . В спектрометрического массовой визуализации , срезы ткани анализируют непосредственно с помощью MALDI-TOF- типа масс - спектрометра .
Чтобы упростить анализ, извлечение часто выполняется путем исключения посттрансляционных модификаций . Сохраняется только первичная структура белков, то есть их последовательность. Но если предметом анализа является изучение этих посттрансляционных модификаций , следует принять необходимые меры для их сохранения.
Разделение
Второй этап позволяет разделить белки в соответствии с их физическими или химическими характеристиками или в соответствии с их сродством к лиганду.
Электрофорез отделяет белки в полиакриламидном геле в соответствии с их молекулярной массы , когда подвергаются действию электрического поля. Стандартный метод электрофореза для протеомики - это двумерный электрофорез .
Хроматографии используют разницу в аффинности белка между подвижной фазой и стационарной фазой.
Принцип разделения белков с помощью двумерного электрофореза
Его принцип состоит в том, что сначала проводят разделение белков по их заряду ( изоэлектрическое фокусирование ) с последующим ортогональным разделением в соответствии с их молекулярной массой. Разрешение по первому измерению составляет порядка 0,01 единицы pH.
Затем полученные гели окрашивают и оцифровывают. Результат - полуквантование.
Поиск интересующих белков с помощью анализа изображений
Сегодня существует два основных подхода, начиная с анализа изображений гелей 2-DE, что позволяет заняться количественной протеомикой. В одном из этих методов используется статистическое сравнение нескольких гелей, а в другом - химический метод получения белков с помощью флуоресцентных зондов, позволяющий проводить комбинированный анализ нескольких образцов на одном геле. Для этого в обоих случаях используется программное обеспечение для обработки изображений. Действительно, невозможно индивидуально воспринять значительное количество пятен (иногда до 2000), рассосавшихся на 2D-геле (рис. 5). Эти множественные пятна соответствуют изоформам белков, разделенных в двух измерениях. Положение полипептидных пятен на геле воспроизводимо в системах градиентного разделения иммобилина. Таким образом, изменение положения является индикатором посттрансляционной модификации, влияющей на его нагрузку и / или размер.
Анализ изображений основан на оцифровке изображения геля 2-DE после окрашивания. На этом этапе программное обеспечение разрезает изображение на пиксели (сжатие элемента изображения) для передачи и хранения данных. Каждый пиксель изображения записывается в положениях x и y, связанных со значением оптической плотности (OD), пропорциональным интенсивности сигнала, записанного камерой или сканером. Чтобы OD была хорошим параметром измерения и отражала экспрессию белка, применяемое окрашивание должно иметь большой динамический диапазон и, если возможно, линейное. В геле, с точки зрения ОП, отношение интенсивности между наименьшим обнаруживаемым пятном и наибольшим пятном составляет порядка 104, в то время как динамика экспрессии белков в клетке составляет от 105 до 106. аналитический диапазон, который необходимо учитывать при анализе.
Окрашивание белков в геле 2-DE основано в основном на использовании органических красителей, таких как синий кумасси , металлов, таких как нитрат серебра, или с помощью флуоресцентных зондов. Диапазон обнаружения варьируется примерно в 10 000 раз между методами с использованием кумасси синего (обнаружение пятен, содержащих количество белка порядка мкг) и методами с использованием нитрата серебра, которые позволяют достичь 0,1 нг. Флуоресцентные красители менее чувствительны, чем нитрат серебра, однако обладают большей воспроизводимостью и динамическим диапазоном.
Текущее программное обеспечение для анализа изображений включает элементы трехмерной визуализации пятен геля, позволяющие изменять углы x, y и z, что чрезвычайно полезно для разделения близлежащих пятен. Таким образом, количественная оценка улучшается. Использование таких инструментов позволяет значительно расширить узкое место, связанное с анализом гелей. Однако для надежного дифференциального анализа необходимо сравнение серий, по крайней мере, из трех-четырех гелей. Статистические тесты, такие как эвристический анализ или анализ соответствия, объективно позволяют определить дисперсию между гелями различных экспериментальных серий.
Умножение двумерных гелей, необходимое для получения статистически надежной дифференциальной количественной оценки, однако, является препятствием для высокопроизводительных анализов, для которых интересен метод одного геля. Дифференциальный анализ на одном геле: (технология DIGE для дифференциального гель-электрофореза) была представлена на рынке компанией GE (ранее) Amersham Biosciences. Принцип основан на ковалентной маркировке с использованием флуоресцентных цианинов (например, Cy2, Cy3 и Cy5) белков, содержащихся в двух экстрактах, подлежащих анализу. Доступны три структуры с разными спектрами флуоресценции. Они также имеют группу сложного эфира N-гидроксисукцинимидила, которая делает возможным образование амида посредством реакции нуклеофильного замещения с аминогруппой в эпсилон лизинов белков. Анализ изображений геля DIGE проще, поскольку два образца мигрировали на одном и том же геле. Изображения, полученные на двух длинах волн, накладываются друг на друга и количественно сравниваются с использованием подходящего программного обеспечения с добавлением внутреннего эталона. Двухцветное сравнительное исследование приводит к демонстрации белков, которые отличаются или идентичны в двух образцах. Возможность наличия внутреннего стандарта увеличивает надежность количественных измерений. Независимо от используемого метода анализа, после обнаружения представляющие интерес пятна вырезаются из геля, чтобы их можно было идентифицировать спектрометрическими методами (масс-спектрометрия в режиме MALDI-TOF или в режиме тандемной МС / МС). В дополнение к дифференциальному анализу, анализ изображений позволяет создавать и аннотировать справочные карты, служащие основой для баз данных, к которым можно обращаться через WEB.
Идентифицировать, охарактеризовать и количественно определить белки
- Фактическая масса белков часто измеряются с помощью масс - спектрометрии , с точностью в пределах от 0,1 дальтона до 10 дальтонов; Переваривание белка ферментом, таким как трипсин, дает фрагменты определенного размера. Затем массу фрагментов измеряют масс-спектрометрией (метод снятия отпечатков пальцев ).
- Методом тандемной масс-спектрометрии и секвенирования по Эдману можно секвенировать пептиды. Но эти методы рассматривают белки как фиксированные структуры, пока они движутся, и что они иногда могут ненадолго связываться с реактивными веществами, связываясь с ними, прежде чем отделиться от них.
- Новый метод дает более точную информацию о пространственной структуре белков, изучая их в растворе и комбинируя известные методы, включая классический рентгеновский или ЯМР-анализ, с ЯМР- спектроскопией, связанной с новым программным обеспечением для анализа, которое обеспечивает доступ к тонким атомным деталям пространственная структура. Нитроксильные группы с неспаренным электроном вводятся в сам белок. Они используются для измерения различий между субъединицами, а затем для определения трехмерной структуры белка или нескольких связанных белков (белковых комплексов, даже больших размеров). Это позволяет лучше изучить связи белков с их партнерами и вывести определенные сложные механизмы биологической регуляции.
Идентификация масс-спектрометрией (МС)
Идентификация с помощью МС основана на точном измерении массы ионизированных пептидов. В большинстве случаев белки перевариваются эндопептидазой (чаще всего трипсином), а затем анализируются с помощью SM.
Проблема решается путем частичного секвенирования белков с помощью тандемной масс-спектрометрии (МС / МС). Затем выбирают и фрагментируют определенные пептидные фрагменты, проанализированные во время первого МС. Полученные массовые пики представляют собой представление белковой последовательности, в которой два соседних пика отличаются массой аминокислоты, потерянной во время фрагментации анализируемого пептида. Тогда аналогию с короткой последовательностью белка можно будет искать в базах данных. Если эти последовательности являются общими для группы белков, изоэлектрическая точка и кажущаяся масса, определенные во время разделения с помощью электрофореза, позволяют решить. Информация о перекрестной проверке (короткие последовательности нескольких аминокислот, расположение окна, содержащего пятно в 2D-электрофорезе, виды животных и тип клеток, из которых взят образец) повышает надежность идентификации полипептида.
Вместо того , чтобы просто определить пептидную последовательность из масс - спектра пептида и использовать его для шахты базы данных белков или ДНК, спектр МСА / МСА можно сравнить с серией спектров MS. / Виртуальный МС , полученный из белковых последовательностей из баз данных . По тому же принципу сначала можно разделить пептиды, полученные в результате триптического расщепления, нанометодом жидкостной хроматографии (наноЖХ) и провести МС / МС этих различных пептидов. Умножение информации о различных сегментах белка затем позволит не только закрепить идентификацию, но и получить структурную информацию, в частности, о посттрансляционных модификациях, таких как добавление фосфатных групп (фосфорилирование) или олигосахаридных цепей. (гликозилирование). С функциональной точки зрения эта информация является фундаментальной. Фосфорилирование является основой передачи сигналов в клетке извне посредством ее мембранных рецепторов к ядру, где централизована информация, регулирующая жизнь клетки. Что касается олигосахаридных цепей, они играют решающую роль в модулировании химических свойств определенных белков (гликопротеинов) и иногда регулируют их биологическую активность. Для идентификации этих групп SM будет использовать фрагменты, полученные в результате их отделения от пептидной цепи или в результате их собственной фрагментации.
Запросы к базам данных
Это последний шаг для протеомика, который стал возможным благодаря достижениям в биоинформатике .
Различная информация, собранная о белках (кажущаяся масса, реальная масса, изоэлектрическая точка, размер фрагментов после ферментативного расщепления, частичные последовательности), сравнивается с геномными или протеомными базами данных в Интернете. Затем программа предоставляет список белков и связанных с ними вероятностей.Количественная оценка экспрессии белка
Это позволяет количественно оценить вариации в уровнях их экспрессии в зависимости от времени, окружающей среды, состояния развития, физиологического и патологического состояния и вида происхождения.
Наиболее часто используемые методы:
Белковые взаимодействия
Она также изучает взаимодействия белков с другими белками, с ДНК или РНК, с веществами.
Другой способ взглянуть на взаимодействия белков - это сделать двойной гибрид . Путем скрининга библиотеки кДНК с ее белком в качестве приманки все интеррактанты могут быть обнаружены в конкретном организме или ткани (животном или растении). При правильном использовании этот прием очень эффективен. Качество результатов часто зависит от качества библиотеки и эффективности трансформации дрожжей или бактерий.
Функциональная протеомика
Функциональная протеомика изучает функции каждого белка.
Структурная протеомика
Протеомика наконец изучает первичную , вторичную и третичную структуры белков.
Вопросы протеомики
Биомаркерные исследования
Новые терапевтические инструменты
Сравнительные исследования киномов раковых клеток позволяют изучать механизмы резистентности и определять новые терапевтические мишени.
Читайте также: