
Презентация на тему фенилкетонурия кратко
Обновлено: 02.07.2024
Свидетельство и скидка на обучение каждому участнику
Зарегистрироваться 15–17 марта 2022 г.

Описание презентации по отдельным слайдам:
Фенилкетонурия Автор: Зыкова О.С , учитель коррекционных классов МБОУ СШ №2

Фенилкетонурия — наследственное заболевание, связанное с нарушением аминокислотного обмена, обусловленное недостаточностью печеночных ферментов, участвующих в метаболизме фенилаланина до тирозина, с последующим тяжелым поражением ЦНС проявляющимся, в частности, в виде нарушения умственного развития. Это одно из немногих наследственных заболеваний, поддающихся успешному лечению.

Причины заболевания Фенилкетонурия является заболеванием с аутосомно-рецессивным характером наследования. Это означает, что для развития клинических признаков фенилкетонурии ребенок должен унаследовать по одной дефектной копии гена от обоих родителей, являющихся гетерозиготными носителями мутантного гена. Фенилкетонурия может быть спровоцирована следующими факторами: Близкородственные браки, при которых, помимо иных патологий, повышается вероятность рождения ребенка с этим заболеванием; Мутация гена (т.е. его изменение), произошедшая по тем или иным причинам в области локализации 12 хромосомы.

Клиническая картина Новорожденные с фенилкетонурией не имеют клинических признаков заболевания. Дети, страдающие фенилкетонурией, рождаются с правильно сформированным и нормально функционирующим головным мозгом. Однако при фенилкетонурии биохимические нарушения головного мозга грудничка начинаются сразу же после рождения. Обычно манифестация фенилкетонурии у детей происходит в возрасте 2-6 месяцев.

Клиническая картина С началом кормления в организм ребенка начинает поступать белок грудного молока либо его заменителей, что приводит к развитию первых, неспецифических симптомов – вялости, иногда – беспокойства и гипервозбудимости, срыгивания, мышечной дистонии, судорожного синдрома. Одним из ранних патогномоничных признаков фенилкетонурии служит упорная рвота, которая нередко ошибочно расценивается как проявление пилоростеноза.


Клиническая картина У детей с фенилкетонурией, не получающих лечения, выявляется микроцефалия (уменьшение размеров черепа и головного мозга), прогнатия, позднее (после 1,5 лет) прорезывание зубов, гипоплазия эмали. Отмечается задержка речевого развития, а к 3-4 годам выявляется глубокая олигофрения и практически полное отсутствие речи.


Клиническая картина Клинические проявления фенилкетонурии II типа характеризуются тяжелой степенью умственной отсталости, повышенной возбудимостью, судорогами, спастическим тетрапарезом (снижение двигательной активности рук и ног), сухожильной гиперрефлексией. Прогрессирование заболевание может приводить к гибели ребенка в возрасте 2-З лет. При фенилкетонури III типа развивается триада признаков: микроцефалия (уменьшение размеров черепа), олигофрения, спастический тетрапарез

Лечение Главным способом лечени является диетотерапия, ограничивающая поступление в организм пищевого белка и фенилаланина до минимальной возрастной потребности. В пищевой рацион больных входят овощи, фрукты, соки, а также специальные малобелковые продукты (саго, хлеб, вермишель, крупка, приготовленная на крахмальной основе).

Лечение Однако в период интенсивного роста и развития ребенка поступление белка в организм должно быть достаточным, так как дефицит его незамедлительно отразится на процессе формирования всех органов и систем. Поэтому из рациона новорожденного нельзя полностью исключить материнское молоко. Для коррекции питания детям даются белковые гидролизаты, лишенные фенилаланина, но содержащие все другие необходимые аминокислоты. Больные нуждаются в дополнительном введении витаминов, особенно группы В, минеральных веществ и микроэлементов.

Методика обучения и воспитания детей с фенилкетонурией В основе занятий с такими детьми должен лежать комплекс коррекционно-развивающих и психологических приемов, позволяющих преодолеть недоразвитие речи и нарушения познавательной деятельности, недостатки эмоционально-волевой сферы, поведения и негативных черт личности. Необходимо развивать у детей высшие психические функции, формировать познавательный интерес, целенаправленность и настойчивость в деятельности. Без этого невозможны реализация их интеллектуальных возможностей и адаптация в социальной среде.

Методика обучения и воспитания детей с фенилкетонурией Огромную роль в реабилитации больных ФКУ играет логопедическое воздействие. С момента поступления больных в отделение логопед в течение двух недель проводит совместно с врачом-психиатром диагностику речевых и интеллектуальных нарушений у детей, составляет перспективный план коррекционной работы, подбирает методы и приемы обучения строго в индивидуальном порядке.

Методика обучения и воспитания детей с фенилкетонурией Затем в соответствии с планом и расписанием в течение всего срока госпитализации с детьми проводятся ежедневные индивидуальные и 2-3 раза в неделю подгрупповые занятия. С детьми старшего дошкольного возраста параллельно с педагогом логопед ведет подготовку к обучению в школе.

Методика обучения и воспитания детей с фенилкетонурией 1. Установление контакта. Например, с помощью педагога ребенок выполняет несложный рисунок (яблоко, цветок, солнце) и всегда получает похвалу. Кроме того, в конце каждого занятия проводится короткая и интересная для ребенка игра (по его выбору), которая назначается как бы за хорошее поведение на занятии. Через некоторое время (обычно через 1-2 мес.) у ребенка возникает устойчивое положительное отношение к занятиям, появляется желание посмотреть на педагога, чтобы увидеть его реакцию на свои действия, устанавливается рабочий контакт с ним.

Методика обучения и воспитания детей с фенилкетонурией 2. Введение оценки деятельности. В беседе ребенка знакомят со школьными оценками - "отлично" ("5") и "плохо" ("2"). "Пятерку" всегда пишут красным цветом, а "двойку" - черным, что придает им особую аффективную значимость. Сначала оцениваются только легкие задания, с которыми ребенок хорошо справляется, например рисование простых форм или предметов. Вскоре детей уже не удовлетворяет устная похвала за проделанную работу, и педагог выставляет им "школьную" отметку. Важнейшей задачей педагога будет подготовка ребенка к преодолению ситуации неуспеха.

Методика обучения и воспитания детей с фенилкетонурией 3. Развитие самоконтроля. Детям предлагают проверить работы других учеников (списывание слов из букваря печатными буквами), в которых либо были допущены ошибки, либо ошибки отсутствовали. Детям говорят, что они должны выставить оценку за работу - "пять" или "два" или оставить работу без оценки. Это задание выполняется с большой охотой - дети проверяют по букварю написание слов (Маша, рама, ушла) и ставят оценки. Затем предлагается оценивать так свою работу, не случается, чтобы ребенок поставил себе плохую оценку. Но спустя некоторое время ребенок уже начинает адекватно оценивать работу

Методика обучения и воспитания детей с фенилкетонурией 4. Развитие самооценки. Как следствие проделываемой в течение первого полугодия работы у детей начинает формироваться адекватная самооценка в сфере учебных действий и заданий (о самооценке личностных качеств ниже). Это проявляется, прежде всего, в том, как они выполняют задание на исследование уровня притязаний.

Методика обучения и воспитания детей с фенилкетонурией 5. Преодоление эгоцентризма. Работа в направлении преодоления эгоцентризма начинается с чтения и обсуждения рассказов, которые требуют оценки ситуации и сопереживания персонажу. Примерами таких рассказов являются "Горькое лекарство" и "Самая красивая". Итогом такой работы становится то, что к концу года дети становятся внимательнее и добрее к родным, менее холодны. Они уже различают состояние и настроение матери (усталая, грустная, веселая) и учитывают это в своих поступках. Отношения с родителями становятся более теплыми.

Методика обучения и воспитания детей с фенилкетонурией 6. Изменение родительских установок на воспитание ребенка. Присутствуя на занятиях, выполняя с ребенком домашние задания, родители начинают видеть его как бы с другой стороны. Если раньше они считали своей основной задачей лечение ребенка, то теперь начинают понимать, что одного лечения недостаточно. Ребенка нужно учить, так как он отстает от сверстников. Его нужно воспитывать, поскольку, если даже делать скидку на болезнь, его личность имеет целый ряд негативных черт. Совместные занятия, игры ведут и к лучшему взаимопониманию родителей и ребенка. Таким образом, меняется место ребенка в семье - на него начинают смотреть не как на больного, а как на будущего ученика
Краткое описание документа:
Фенилкетонурия – наследственное нарушение аминокислотного обмена, обусловленное недостаточностью печеночных ферментов, участвующих в метаболизме фенилаланина до тирозина. Ранними признаками фенилкетонурии служат рвота, вялость или гиперактивность, запах плесени от мочи и кожи, задержка психомоторного развития; типичные поздние признаки включают олигофрению, отставание в физическом развитии, судороги, экзематозные изменения кожи и др. Скрининг новорожденных на фенилкетонурию проводится еще в родильном доме; последующая диагностика включает молекулярно-генетическое тестирование, определение концентрации фенилаланина в крови, биохимический анализ мочи, ЭЭГ, МРТ головного мозга. Лечение фенилкетонурии заключается в соблюдении специальной диеты.
Вы можете изучить и скачать доклад-презентацию на тему Фенилкетонурия. Презентация на заданную тему содержит 13 слайдов. Для просмотра воспользуйтесь проигрывателем, если материал оказался полезным для Вас - поделитесь им с друзьями с помощью социальных кнопок и добавьте наш сайт презентаций в закладки!


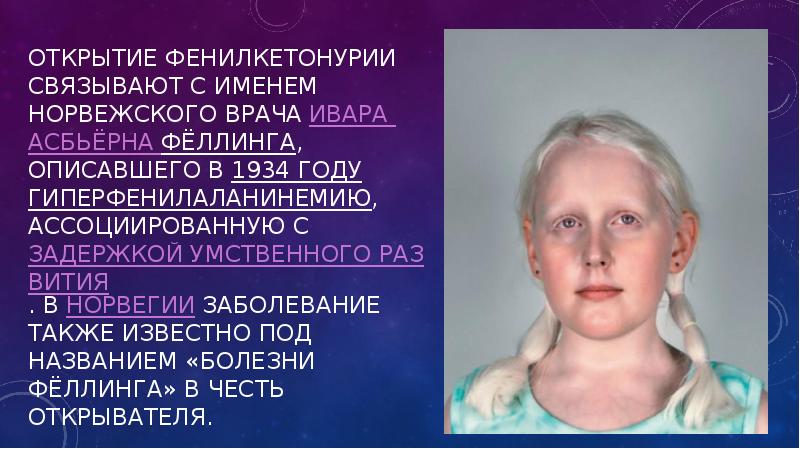










Фенилкетонури́я — наследственное заболевание группы ферментопатий, связанное с нарушением метаболизма аминокислот, главным образом фенилаланина. При несоблюдении низкобелковой диеты сопровождается накоплением фенилаланина и его токсических продуктов, что приводит к тяжёлому поражению ЦНС, проявляющемуся, в частности, в виде нарушения умственного развития (фенилпировиноградной олигофрении).
Фенилпировиноградная олигофрения — наследственное заболевание из числа нарушений обмена аминокислот. Встречается у новорожденных с частотой 1:10000. Дети с этой болезнью составляют до 12% среди умственно отсталых.
В основе заболевания лежит снижение активности или полное отсутствие фермента фенилаланингидроксилазы, участвующего в обмене фенилаланина. Вследствие дефекта обмена этой аминокислоты происходит приводящее к поражению нервной системы избыточное накопления токсических веществ (кетокислоты).
Ведущим клиническим симптомом болезни является отставание в психическом развитии, которое может быть выражено в различной степени. Снижение интеллекта сочетается с отставанием в физическом развитии: больные дети поздно начинают сидеть, стоять, ходить. Рост больных обычно ниже нормального.
В ранней стадии болезни снижается тонус мышц, затем постепенно формируются спастические парезы и параличи. Сухожильные рефлексы обычно высокие, с расширенными зонами. Походка спастико-атактическая вследствие высокого мышечного тонуса и нарушений координации. Нередко уже в первые годы у больных отмечаются судороги.
Неврологические расстройства также могут проявляться в форме постепенного нарастания непроизвольных движений — гипер-кинезов. Наблюдаются вращательные движения туловища или отдельных частей тела, червеобразные сокращения мышц пальцев (атетоз), дрожание пальцев вытянутых рук (тремор). Изменения черепных нервов выражаются в нарастающем косоглазии и нистагме.
У большинства нелеченых больных снижение интеллекта постепенно прогрессирует до степени идиотии. Характерны расстройства речи или нарушения ее формирования. У большинства больных речь полностью отсутствует. У других детей она бедна, односложна, часто аграмматична, у них же отмечаются эхолалия и персеверации. Навыки опрятности, самообслуживания обычно Формируются с трудом. Выражены нарушения эмоциональной сферы.
Лечение фенилкетонурии проводится диетотерапией - необходимо придерживаться диеты со строгими ограничениями содержания в продуктах фенилаланина из-за того, что эта аминокислота в огромном количестве есть в белке, из рациона абсолютно исключаются вся белковая пища животного происхождения - это, молоко, мясо, рыба, грибы и прочее.
Строжайшая диета обязана соблюдаться как минимум в течение пяти лет жизни. В более взрослом возрасте значительно снижается восприимчивость нервной системой опасному воздействию фенилаланина и его продуктов распада. Практически все детки в возрасте 12-14 лет могут свободно переходить на обычное питание.

№ слайда 1
Фенилкетонурия ФКУ финилпировиноградная олигофрения болезнь Фёллинга

№ слайда 2
Фенилкетонурия Наследственное заболевание обмена одной из важных аминокислот (фенилаланина), в связи с недостатком или полным отсутствием необходимого для обмена фермента. Это приводит к накоплению в организме особо токсичных веществ, поражающих нервную систему.

№ слайда 3
Местоположение гена в хромосоме (локус): 12 q 22 – 24, 2 4 р 15,3
№ слайда 4
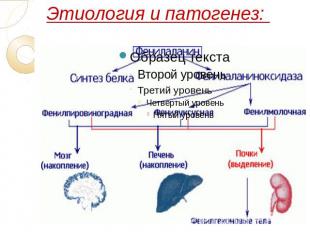
Этиология и патогенез:
№ слайда 5

№ слайда 6
Психопатологически отмечается: Умственная отсталость( 65% - глубокая, 31.8% - умеренная и 3.2% - легкая)Недоразвитие речи (ее или совсем нет, или есть отдельные слова, которые больные не соотносят с объектом), т.е. нарушено: - понимание речи - звукопроизношение
№ слайда 7
Неврологическая симптоматика: Эпилептиформные припадкиНарушение мышечного тонусаПлохая координация движенийМного стереотипии, часты другие знаки экстрапирамидной недостаточности (атетоидные, хореиформные движения)

№ слайда 8
Расстройства поведения: Двигательное беспокойство, целенаправленные, неуправляемые перемещения от объекта к объекту, бесцельные манипуляции с предметами.илиДети пассивны, вялы, плохо узнают близких, оживляются при упоминании о еде.

№ слайда 9
Патологоанатомически обнаруживается: - малая масса мозга - дефекты миелинезации в коре больших полушарии( особенно в лобных и височных долях), и других структурах (внутренняя капсула, зрительные проводящие пути) - депигментация черной субстанции

№ слайда 10
Обследование детей: Все новорожденные обследуются по специальным программам скрининга на повышение концентрации фенилаланина. Используют: 1.Микробиологический метод определения концентрации фенилаланина в крови.2. Проба Фёллинга на фенилпировидноградную кислоту в моче (берется 5% раствор треххлористого железа и уксусной кислоты и прибавляется несколько капель к моче, появление зеленой окраски говорит о положительной реакции на фенилаланин).В дальнейшем определяют количественное содержание фенилаланина в крови и моче(хроматография аминокислот), , это исследование с помощью специальных анализаторов.

№ слайда 11
Наследственность: Болезнь наследуется по рецессивному типу (т.е. болеют сестры и братья из одной семьи, а родители здоровы, хотя и являются гетерозиготными носителями гена ФКУ). Ген фенилкетонурии встречается в среднем у 1-2 на 100 человек, но болезнь может возникнуть лишь в том случае, если и мать и отец ребенка являются носителями этого гена, и ребенок унаследует его в двойном наборе. Поэтому болезнь встречается значительно реже, чем распространен ген. Больные ФКУ (обладатели двух патологических генов) могут иметь детей с фенилкетонурией только при вступлении в брак с носителями таких же генов. При вступлении в брак с лицами свободными от гена ФКУ, дети не болеют этим заболеванием.

№ слайда 12
Диетотерапия: Исключить: мясо, колбасы, рыбу, бульоны, яйца, творог, сыр, мучные изделия, каши из естественных круп, фасоль, орехи, шоколад. Меню для детей составляется из: фруктов, овощей, крахмальных изделий, жиров, со строгим учетом содержания в них фенилаланина.

№ слайда 17
При несоблюдении женщиной специфической диеты во время беременности у ребёнка возникают следующие пороки развития: в 92% случаев - умственная отсталость в 73% случаев - микроцефалия в 12% случаев - врождённые пороки сердца в 40% случаев - низкая масса тела при рождении

№ слайда 18

№ слайда 19
Работу выполнили: Панкратова В.С.Сухарева М.В.Сионская Н.В.Зубанова Н.А.Богачева А.Н.Чилипалова И.СКиселева М.Е.Святохо И.А.Белькова Я.В.Петрова Н.Л.
Фенилкетонурия – это наследственное нарушение аминокислотного обмена, обусловленное недостаточностью печеночных ферментов, участвующих в метаболизме фенилаланина до тирозина. Ранними признаками фенилкетонурии служат рвота, вялость или гиперактивность, запах плесени от мочи и кожи, задержка психомоторного развития; типичные поздние признаки включают олигофрению, отставание в физическом развитии, судороги, экзематозные изменения кожи и др. Скрининг новорожденных на фенилкетонурию проводится еще в родильном доме; последующая диагностика включает молекулярно-генетическое тестирование, определение концентрации фенилаланина в крови, биохимический анализ мочи, ЭЭГ, МРТ головного мозга. Лечение фенилкетонурии заключается в соблюдении специальной диеты.
МКБ-10
Общие сведения
Фенилкетонурия (болезнь Феллинга, фенилпировиноградная олигофрения) – врожденная, генетически обусловленная патология, характеризующаяся нарушением гидроксилирования фенилаланина, накоплением аминокислоты и ее метаболитов в физиологических жидкостях и тканях с последующим тяжелым поражением ЦНС. Фенилкетонурия впервые описана А. Феллингом в 1934 г.; встречается с частотой 1 случай на 10 000 новорожденных.
В неонатальном периоде фенилкетонурия не имеет клинических проявлений, однако поступление фенилаланина с пищей вызывает манифестацию заболевания уже в первом полугодии жизни, а в дальнейшем приводит к тяжелым нарушениям развития ребенка. Именно поэтому пресимптоматическое выявление фенилкетонурии у новорожденных является важнейшей задачей неонатологии, педиатрии и генетики.
Причины фенилкетонурии
Фенилкетонурия является заболеванием с аутосомно-рецессивным характером наследования. Это означает, что для развития клинических признаков фенилкетонурии ребенок должен унаследовать по одной дефектной копии гена от обоих родителей, являющихся гетерозиготными носителями мутантного гена.
Чаще всего к развитию фенилкетонурии приводит мутация гена, кодирующего фермент фенилаланин-4-гидроксилазу и расположенного на длинном плече 12 хромосомы (локус12q22-q24.1). Это, так называемая, классическая фенилкетонурия I типа, составляющая 98% всех случаев заболевания. Гиперфенилаланинемия может достигать 30 мг% и выше. При отсутствии лечения данный вариант фенилкетонурии сопровождается глубокой умственной отсталостью.
Кроме классической формы, различают атипичные варианты фенилкетонурии, протекающие с той же клинической симптоматикой, но не поддающиеся коррекции диетотерапией. К ним относятся фенилкетонурия II типа (недостаточность дегидроптеринредуктазы), фенилкетонурия III типа (дефицит тетрагидробиоптерина) и другие, более редкие варианты. Вероятность рождения ребенка, больного фенилкетонурией, повышается при заключении близкородственных браков.
Патогенез
В основе классической формы фенилкетонурии лежит недостаточность фермента фенилаланин-4-гидроксилазы, участвующего в конверсии фенилаланина в тирозин в митохондриях гепатоцитов. В свою очередь, производный тирозина – тирамин является исходным продуктом для синтеза катехоламинов (адреналина и норадреналина), а дийодтирозин – для образования тироксина. Кроме этого, результатом метаболизма фенилаланина служит образование пигмента меланина.
Наследственная недостаточность фермента фенилалаиин-4-гидроксилазы при фенилкетонурии приводит к нарушению окисления фенилаланина, поступающего с пищей, в результате чего его концентрация в крови (фенилаланинемия) и спинномозговой жидкости значительно возрастает, а уровень тирозина соответственно падает. Избыточное содержание фенилаланина устраняется путем повышенной экскреции с мочой его метаболитов - фенилпировиноградной, фенилмолочной и фенилуксусной кислот.
Нарушение обмена аминокислот сопровождается нарушением миелинизации нервных волокон, снижением образования нейромедиаторов (дофамина, серотонина и др.), запускающими патогенетические механизмы задержки умственного развития и прогредиентное слабоумие.
Симптомы фенилкетонурии
Новорожденные с фенилкетонурией не имеют клинических признаков заболевания. Обычно манифестация фенилкетонурии у детей происходит в возрасте 2-6 месяцев. С началом кормления в организм ребенка начинает поступать белок грудного молока либо его заменителей, что приводит к развитию первых, неспецифических симптомов – вялости, иногда – беспокойства и гипервозбудимости, срыгивания, мышечной дистонии, судорожного синдрома. Одним из ранних патогномоничных признаков фенилкетонурии служит упорная рвота, которая нередко ошибочно расценивается как проявление пилоростеноза.
У детей с фенилкетонурией, не получающих лечения, выявляется микроцефалия, прогнатия, позднее (после 1,5 лет) прорезывание зубов, гипоплазия эмали. Отмечается задержка речевого развития, а к 3-4 годам выявляется глубокая олигофрения (идиотия) и практически полное отсутствие речи.
Клинические проявления фенилкетонурии II типа характеризуются тяжелой степенью умственной отсталости, повышенной возбудимостью, судорогами, спастическим тетрапарезом, сухожильной гиперрефлексией. Прогрессирование заболевание может приводить к гибели ребенка в возрасте 2-З лет. При фенилкетонури III типа развивается триада признаков: микроцефалия, олигофрения, спастический тетрапарез.
Диагностика
В настоящее время диагностика фенилкетонурии (а также галактоземии, врожденного гипотиреоза, адрено-генитального синдрома и муковисцидоза) входит в программу неонатального скрининга, осуществляемого всем новорожденным. Основные и дополнительные методы диагностики:
- Скрининг-тест. Проводится на 3-5 день жизни доношенного и 7 день жизни недоношенного ребенка путем забора образца капиллярной крови на специальный бумажный бланк. При обнаружении гиперфенилаланемии более 2,2 мг% ребенка направляют к детскому генетику для повторного обследования.
- Биохимические исследования. Для подтверждения диагноза фенилкетонурии проверяется концентрация фенилаланина и тирозина в крови, определяют активность печеночных ферментов (фенилаланингидроксилазы), выполняется биохимическое исследование мочи (определение кетоновых кислот), метаболитов катехоламинов в моче и др.
- Неврологическое обследование. Дополнительно проводится ЭЭГ и МРТ головного мозга, осмотр ребенка детским неврологом.
- Пренатальная диагностика. Генетический дефект при фенилкетонурии может быть обнаружен еще на этапе беременности в ходе инвазивной пренатальной диагностики плода (хорионбиопсии, амниоцентеза, кордоцентеза). В остальных случаях окончательный диагноз выставляется по результатам ДНК-диагностики после рождения.
Дифференциальный диагноз фенилкетонурии проводят с внутричерепной родовой травмой новорожденных, внутриутробными инфекциями, другими нарушениями обмена аминокислот.
Лечение фенилкетонурии
Основополагающим фактором в лечении фенилкетонурии является соблюдение диеты, ограничивающей поступление белка в организм. Лечение рекомендуется начинать при концентрации фенилаланина >6 мг%. Для грудных детей разработаны специальные смеси - Афенилак, Лофенилак; для детей старше 1 года – Тетрафен, Фенил-фри; старше 8 лет - Максамум-ХР и др. Основу диеты составляют низкобелковые продукты - фрукты, овощи, соки, белковые гидролизаты и аминокислотные смеси. Расширение диеты возможно после 18 лет в связи с возрастанием толерантности к фенилаланину. В соответствии с российским законодательством обеспечение лиц, страдающих фенилкетонурией, лечебным питанием, должна осуществляться бесплатно.
Больным назначается прием минеральных соединений, витаминов группы В и др.; по показаниям - ноотропные средства, антиконвульсанты. В комплексной терапии фенилкетонурии широко используется общий массаж, ЛФК, иглорефлексотерапия. Атипичные формы фенилкетонурии, не поддающиеся лечению диетой, требуют назначения гепатопротекторов, противосудорожных средств, заместительной терапии леводопой, 5-гидрокситриптофаном.
Дети, страдающие фенилкетонурией, находятся под наблюдением участкового педиатра и психоневролога; нередко нуждаются в помощи логопеда и дефектолога. Необходим тщательный мониторинг нервно-психического статуса детей, контроль уровня фенилаланина в крови и показателей электроэнцефалограммы.
Прогноз и профилактика
Проведения массового скрининга на фенилкетонурию в неонатальном периоде позволяет организовать раннюю диетотерапию и предотвратить тяжелые церебральные повреждения, нарушения функции печени. При раннем назначении элиминационной диеты при классической фенилкетонурии прогноз развития детей хороший. При поздно начатом лечении прогноз в отношении умственного развития неблагоприятный.
Профилактика осложнений фенилкетонурии заключается в проведении массового скрининга новорожденных, раннего назначения и длительного соблюдения диетического питания.
С целью оценки риска рождения ребенка с фенилкетонурией предварительное генетическое консультирование должны пройти супружеские пары, уже имеющие больного ребенка, состоящие в кровнородственном браке, имеющие родственников с данным заболеванием. Женщины с фенилкетонурией, планирующие беременность, должны соблюдать строгую диету до зачатия и во время беременности для исключения повышения уровня фенилаланина и его метаболитов и нарушения развития генетически здорового плода. Риск рождения ребенка с фенилкетонурией у родителей-носителей дефектного гена, составляет 1:4.
Читайте также:
- Памятка для родителей в детском саду по безопасности в осенне зимний период
- Сценарий непослушные котята мюзикл в детском саду
- Относимость и допустимость доказательств в арбитражном процессе кратко
- Г м дульнев вклад в специальную психологию кратко
- Общие принципы геодезических разбивочных работ кратко