
Синдром тестикулярной феминизации доклад
Обновлено: 17.05.2024
Синдром нечувствительности к андрогенам, или синдром тестикулярной феминизации (300068, ген AR рецептора андрогенов [313700], Xq11–q12, À , рецессив]), выявляют у пациенток с мужским кариотипом (46,ХY), но женским фенотипом, половые органы сформированы по женскому типу. Частота тестикулярной феминизации — 1:65 000 генетических мужчин.
Этиология и патогенез • Синдром тестикулярной феминизации (синдром Морриса) — тип мужского псевдогермафродитизма: наличие наружных женских половых органов, недоразвитие репродуктивных органов, вторичных половых признаков и аменорея; образуются как андрогены, так и эстрогены, но рецепторы в значительной степени рефрактерны к андрогенам; у больных нет полового хроматина, они имеют нормальный мужской кариотип • Обычно у новорождённых мальчиков содержание тестостерона повышено в течение нескольких месяцев. Содержание тестостерона, превышающее нормальные показатели, при одновременном увеличении содержания ЛГ может указывать на резистентность к андрогенам либо на нарушение отрицательной обратной связи (ингибирование синтеза тестостерона). Это соотношение сохраняется и у взрослых.
Клиническая картина и диагностика
• При полной резистентности к андрогенам ребёнок с генотипом 46,XY (имеющий яички) внешне выглядит как девочка. Ключ к диагностике — обнаружение яичек в паховом канале. В подростковом возрасте таких детей расценивают как девочек с первичной аменореей. Поскольку яички во внутриутробном периоде синтезировали мюллеров ингибирующий фактор, влагалище представляет собой неглубокий и слепо оканчивающийся карман. Если яички не были удалены до пубертата из-за происходящего в них превращения тестостерона в эстроген, развиваются нормальные грудные железы.
• При частичной устойчивости к андрогенам больной с кариотипом XY имеет гениталии переходного типа. Диагноз установить трудно. Поскольку патология наследуется по X-сцепленному рецессивному типу, мать ребёнка должна быть носителем генного дефекта, а половина её детей должны быть либо девочками (XY-генотип, также носители), либо больными мальчиками. Таким образом, в семейном анамнезе надо искать указания на бесплодие или крипторхизм.
МКБ-10 • E34.5 Синдром андрогенной резистентности
Код вставки на сайт
Синдром нечувствительности к андрогенам, или синдром тестикулярной феминизации (300068, ген AR рецептора андрогенов [313700], Xq11–q12, À , рецессив]), выявляют у пациенток с мужским кариотипом (46,ХY), но женским фенотипом, половые органы сформированы по женскому типу. Частота тестикулярной феминизации — 1:65 000 генетических мужчин.
Этиология и патогенез • Синдром тестикулярной феминизации (синдром Морриса) — тип мужского псевдогермафродитизма: наличие наружных женских половых органов, недоразвитие репродуктивных органов, вторичных половых признаков и аменорея; образуются как андрогены, так и эстрогены, но рецепторы в значительной степени рефрактерны к андрогенам; у больных нет полового хроматина, они имеют нормальный мужской кариотип • Обычно у новорождённых мальчиков содержание тестостерона повышено в течение нескольких месяцев. Содержание тестостерона, превышающее нормальные показатели, при одновременном увеличении содержания ЛГ может указывать на резистентность к андрогенам либо на нарушение отрицательной обратной связи (ингибирование синтеза тестостерона). Это соотношение сохраняется и у взрослых.
Клиническая картина и диагностика
• При полной резистентности к андрогенам ребёнок с генотипом 46,XY (имеющий яички) внешне выглядит как девочка. Ключ к диагностике — обнаружение яичек в паховом канале. В подростковом возрасте таких детей расценивают как девочек с первичной аменореей. Поскольку яички во внутриутробном периоде синтезировали мюллеров ингибирующий фактор, влагалище представляет собой неглубокий и слепо оканчивающийся карман. Если яички не были удалены до пубертата из-за происходящего в них превращения тестостерона в эстроген, развиваются нормальные грудные железы.
• При частичной устойчивости к андрогенам больной с кариотипом XY имеет гениталии переходного типа. Диагноз установить трудно. Поскольку патология наследуется по X-сцепленному рецессивному типу, мать ребёнка должна быть носителем генного дефекта, а половина её детей должны быть либо девочками (XY-генотип, также носители), либо больными мальчиками. Таким образом, в семейном анамнезе надо искать указания на бесплодие или крипторхизм.
Что такое синдром Морриса? Причины возникновения, диагностику и методы лечения разберем в статье доктора Литвинова В. В., репродуктолога со стажем в 39 лет.
Над статьей доктора Литвинова В. В. работали литературный редактор Юлия Липовская , научный редактор Сергей Федосов и шеф-редактор Лада Родчанина

Определение болезни. Причины заболевания
Синдром Морриса (синдром тестикулярной феминизации) — это врождённое генетическое заболевание, при котором у людей мужского пола ткани-мишени не чувствительны к мужским половым гормонам — андрогенам. Человек с синдромом Морриса генетически является мужчиной (имеет кариотип 46 XY), но выглядит как женщина.
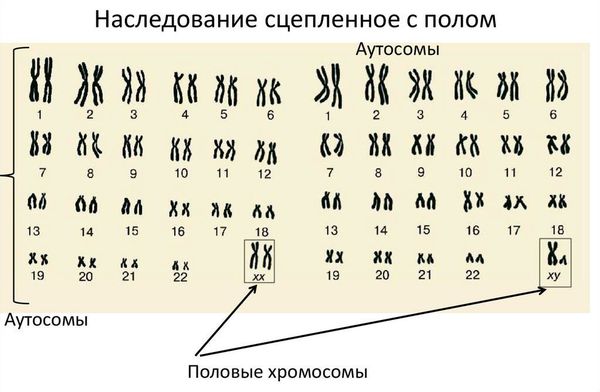
Кариотип — это набор хромосом, который передаётся ребёнку от матери и отца. Кариотип позволяет определить характеристики индивида, включая пол. В норме у человека в генетическом наборе присутствует 46 хромосом, из них 22 пары аутосомных (не определяющих пол) и одна пара половых хромосом, которая определяет гендерную принадлежность ребёнка. Половые хромосомы женщины обозначаются как ХХ, мужчины — ХY. То есть нормальный женский кариотип — 46 XX, мужской — 46 XY.
Причина синдрома Морриса — мутации (изменение) гена рецептора андрогенов, у наследованные от матери или возникшие впервые . Мутации обуславливают резистентность (нечувствительность) рецепторов к гормону тестостерону [19] . В этом случае развивается синдром тестикулярной феминизации (СТФ). Ещё это заболевание называют синдромом Морриса — по имени американского гинеколога, который впервые ввёл термин в обиход, подробно описав его в 1953 году. Другие синонимы патологии — синдром нечувствительности к андрогенам, синдром андрогенной резистентности.
Часто мужчины с синдромом Морриса даже не догадываются о своём биологическом поле (как и их родители) и живут как девочки/женщины. Это объясняется тем, что данная патология больше никак себя не проявляет, кроме проблем с фертильностью (способностью к зачатию) во взрослом возрасте.
В популяции синдром встречается редко. По данным разных авторов, частота заболевания составляет от 1: 20 400 до 1: 99 100 случаев [19] . Точные данные собрать сложно, так как патология часто остаётся нераспознанной. Согласно исследованию, которое проводилось в Дании в течение семилетнего периода, полная нечувствительность к андрогенам случается с вероятностью 1 на 20 400 новорождённых с кариотипом 46 XY [15] .
Синдром тестикулярной феминизации впервые в Европе описан в 1817 году баварским врачом Джорджем Стегленером, в России — в 1893 году профессором клиники московского университета Благоволиным Сергеем Ивановичем (1865-1947) [4] .

Есть заключения генетиков и историков (по утверждению профессора Эфроимсона), которые приписывают синдром тестикулярной феминизации и королеве Англии Елизавете Тюдор (1533-1603) [5] .
Несмотря на то, что синдром Морриса встречается редко, он обнаруживается почти у 1 % выдающихся спортсменок , которые имеют превосходство в физической силе, быстроте и ловкости [5] [18] . Это стало причиной исключения женщин и девушек с синдромом тестикулярной феминизации из женских спортивных состязаний [5] . Регламент многих серьёзных спортивных соревнований, особенно в силовых видах спорта, беге, прыжках, обязывает женщин предоставить результат анализа на кариотип. Обязательное тестирование стали проводить после истории с немецкой легкоатлеткой Дорой Ратьен, которая участвовала в Олимпийских играх 1936 года. После игр стало известно, что Дора генетически была мужчиной. Этот случай послужил поводом к тому, что всех участниц соревнований стали осматривать врачи. Позже вместо осмотров спортсменки стали сдавать анализ крови на кариотип.

Иногда синдром Морриса называют синдромом манекенщиц , так как женщины с данной патологией часто имеют привлекательную внешность [16] [17] .
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы синдрома Морриса
Как правило, люди с синдромом тестикулярной феминизации имеют женский фенотип, т. е. внешне выглядят как женщины. Однако есть некоторые признаки, по которым можно определить наличие заболевания.
- С началом полового созревания больные отличаются женским телосложением. Это объясняется влиянием эстрогенов и отсутствием эффекта тестостерона. П ри этом у таких людей часто высокий рост, узкий таз и широкие плечи, что более характерно для мужчин. [1][2][6][7] .

- При нормальном росте и отсутствии аномалий развития характерны крупные кисти рук и крупные стопы.
- Молочные железы обычно развиты соответственно 3-4 размеру, однако ареолы сосков окрашены бледно.
- Характерно отсутствие полового оволосения (в области подмышечных впадин и на лобке), т. к. "не работает" тестостерон.
- В результате влияния эстрадиола и отсутствия действия тестостерона в ходе эмбрионального развития у плода мужского пола наружные половые органы формируются по женскому типу: не формируется половой член, но есть половые губы, укороченное влагалище, заканчивающееся слепо (т. е. нет матки, шейки матки и яичников).
- У детей с синдромом Морриса (4-5 лет) иногда появляются паховые грыжи. В таких случаях родители обращаются к детскому хирургу. Врач проводит операцию и в составе грыжевого содержимого обнаруживает ткань, напоминающую по структуре яичко (мужскую гонаду). Так как у "девочки" не может быть мужской гонады, берут биопсию (для подтверждения диагноза СТФ). В таком случае яичко не удаляют, ушивают грыжевой мешок и заканчивают операцию. Далее врач рекомендует провести генетическое обследование ребёнка, которое устанавливает мужской кариотип 46 XY. Считается, что удалять гонады до пубертатного периода нельзя, т. к. это может нарушить формирование организма ребёнка в целом.
- В 14-16 лет (если патология не была выявлена раньше), родители замечают, что у "девочки" отсутствует менструация, в связи с чем обращаются к врачу-гинекологу. Во время гинекологического осмотра и УЗИ выявляется, что у ребёнка имеется слепо заканчивающееся влагалище (в виде слепого мешка). Глубина влагалища может варьировать от нормальной до укороченной [1][2][6][7] .
Патогенез синдрома Морриса
В начале эмбрионального развития у зародышей вне зависимости от хромосомного набора, образовавшегося при оплодотворении яйцеклетки сперматозоидом, половая система закладывается одинаково и предоставляет возможности для развития как женской, так и мужской половой системы. В частности, у зародыша одновременно формируются вольфов и мюллеров протоки, которые потом превращаются в семявыносящие протоки у мужчин и матку с фаллопиевыми трубами и влагалищем у женщин. Половые железы эмбриона (гонады) не дифференцированы и содержат первичные половые клетки (гаметы), которые могут превратиться как в клетки яичников (женские гонады), так и в клетки семенников (мужские гонады).
Таким образом, эмбрион до 6 недель является нейтральным по полу (имеет признаки и мужского, и женского пола). Далее процесс формирования половых признаков и в дальнейшем организма происходит под строгим контролем гормонов: у эмбриона мужского пола — под влиянием тестостерона (влагалище атрофируется), у эмбриона женского пола — под влиянием эстрадиола и прогестерона (влагалище трансформируется из "слепого мешка" и формируется шейка матки и матка). Тестостерон и эстрадиол вырабатываются как у мужчин, так и у женщин, только их соотношение разное. В норме у мальчиков соотношение мужского гормона к женскому 4\1, а в случае СТФ — 0\1. У девочек соотношение мужского гормона к женскому, прямо противоположное — 1\4.
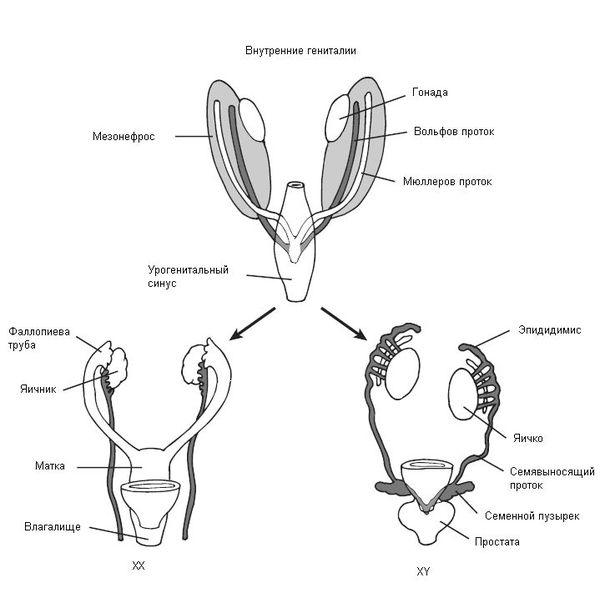
В процессе эмбриогенеза у людей с синдромом Морриса под влиянием мужской Y-хромосомы гонады развиваются как яички, они не способны к сперматогенезу, но способны вырабатывать тестостерон [8] . Однако из-за генетической мутации организм "не видит"/"не чувствует" присутствие тестостерона в крови, поэтому гормон не может проявить свои свойства и сформировать мужской организм. При этом секретируемые надпочечниками эстрогены (эстрадиол), хоть и в небольшом количестве, нормально вырабатываются и усваиваются организмом, в результате чего начинают превалировать в организме (как у девочки). В связи с этим влагалище не атрофируется, в дальнейшем ребёнок развивается по женскому типу.
Классификация и стадии развития синдрома Морриса
Синдром тестикулярной феминизации делят на две формы — полный и неполный. Дети с полной формой нечувствительности к мужским гормонам имеют однозначно женский внешний вид. При этом состоянии чувствительность организма к мужскому половому гормону (тестостерону) отсутствует полностью. Рождается здоровая "девочка", не имеющая, на первый взгляд, каких-либо отклонений в развитии.
Неполная форма СТФ характеризуется более разнообразной клинической картиной. В этом случае отмечается некоторая чувствительность рецепторов к тестостерону. Выделяют пять основных степеней неполного синдрома тестикулярной феминизации (классификация 1996 года) [10] .
- Мужской тип. У больных мужской фенотип без каких-либо отклонений.
- Преимущественно мужской тип. Наблюдаются нарушения формирования половых органов, хотя фенотип больных — мужской.
- Амбивалентный тип. Более выраженные нарушения формирования половых органов: уменьшение полового члена, который становится похожим на клитор, мошонка похожа на большие половые губы. Характерно расширение таза, узкие плечи, гинекомастия (увеличение груди).
- Преимущественно женский тип. По фенотипическим признакам больные являются женщинами. Однако у них, как правило, короткое влагалище и гипертрофированый (увеличенный) клитор
- Женский тип. По всем внешним признакам, за исключением увеличенного клитора, больные являются женщинами.
Осложнения синдрома Морриса
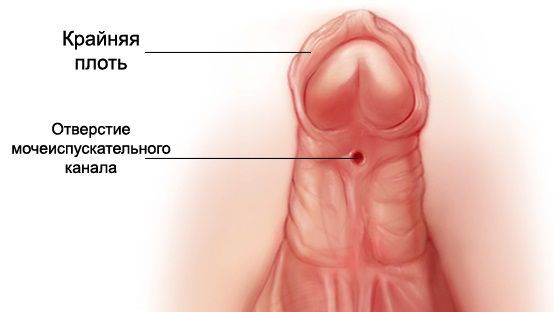
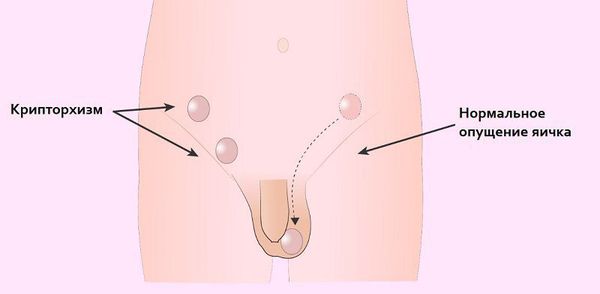
- Из-за нарушения проходимости (опущения) яичек по паховому каналу у больных СТФ в детском возрасте (3-5 лет) часто обнаруживают паховые грыжи и гипоспадию ( недоразвитие полового члена и неправильное расположение мочеиспускательного канала) . Гипоспадия, в свою очередь, может стать причиной развития различных воспалительных процессов в мочевыделительной системе ( уретриты , пиелонефриты) [13] .
- Крипторхизм (неопущение яичек) в будущем грозит злокачественным перерождением тканей яичка (развитием гонадобластомы), что является наиболее тяжёлым осложнением данного заболевания [10][19] . Одна из причин образования гонадобластомы в том, что яички постоянно находятся в забрюшинном пространстве, где температура выше 37 °C. В норме яички должны находиться снаружи (в мошонке), т. к. для их жизнедеятельности нужна постоянная температура ниже 34 °C. Точную частоту возникновения рака у пациентов с синдромом тестикулярной феминизации оценить очень трудно, однако по различным данным общий риск составляет примерно 5 % всех случаев патологии. При этом с возрастом вероятность развития рака возрастает [reference:19 ] .
- Ещё одним серьёзным осложнением заболевания является бесплодие.
Диагностика синдрома Морриса
- Определение фенотипа — совокупности внешних признаков.
- Изучение семейного анамнеза пациента. Если в семье были случаи андрогенной нечувствительности, то вероятность наличия патологии выше, однако нужно понимать, что отсутствие отягощённого семейного анамнеза не исключает диагноз.
- Гинекологический и урологический осмотр. Выявляется слепо заканчивающееся влагалище (слепой мешок), отсутствие шейки, не пальпируется матка и её придатки.
- Определение половых гормонов в крови (тестостерона, эстрадиола). Выявляется высокий уровень тестостерона, уровень эстрадиола выше, чем у мужчин, но ниже, чем у женщин в норме.
- Ультрасонографии органов малого таза (УЗИ) и р ентгенологическое обследование для выяснения состояния органов малого таза . В малом тазу не визуализируется матка и яичники. Забрюшинно визуализируются образования, похожие на "яичники", на самом деле это яички (без дополнительных данных анамнеза бывает трудно это предположить). Мужские гонады могут располагаться в паховых каналах, в стенках таза или в толще больших половых губ.
- Кариотипирование — исследование хромосомного набора. Позволяет обнаружить отклонения в структуре и числе хромосом . При синдроме тестикулярной феминизации определятся мужской кариотип — 46 XY. Для исследования используется венозная кровь.
- Молекулярно-генетический анализ гена андрогенного рецептора. Определение мутаций гена при наличии характерной клинической картины подтверждает диагноз СТФ с вероятностью близкой к 100 %.
Лечение синдрома Морриса
Лечение синдрома тестикулярной феминизации должно осуществляться междисциплинарной командой врачей, которая состоит из хирурга, гинеколога, генетика, эндокринолога и клинического психолога или психиатра.
При полной андрогенной нечувствительности и в случае частичной нечувствительности с преимущественно женским типом в 20-50 % случаев необходимо удалять яички из-за риска развития рака [19] . Операция, как правило, проводится после завершения пубертатного периода и конституционального формирования (14-15 лет) [11] . Хирургическое лечение в отношении половых желёз (гонад) проводится в настоящее время лапароскопическим доступом [3] [6] . Лапароскопию должен проводить хирург высокой квалификации, так как удаляемые гонады (яички) находятся забрюшинно и имеют высокую степень кровоснабжения. Динамическое наблюдение, УЗИ и контроль гормона тестостерона позволяют оценивать эффективность проведённой операции [14] .
Операция эффективна в 100 % случаев. Но так как удаляемые гонады мягкие и не имеют чёткой формы и структуры, возможны случаи неполного их удаления. Тогда оставшаяся ткань яичка может возобновить свою работу, что проявляется повышением тестостерона в крови и визуализацией на УЗИ гонады/гонад. В этом случае необходима повторная операция. Однако это случается очень редко.
После удаления гонад показана последующая длительная заместительная терапия эстрогенами. Она необходима для продолжения дальнейшего формирования женского организма и обязательна (до возраста 45-50 лет) для профилактики остеопороза (потери костной тканью кальция) [8] [12] . Развитие остеопороза связано с тем, что в организме пациента мало собственного эстрадиола, который в норме удерживает кальций в костях. Следовательно, у таких людей кальций будет вымываться быстрее и плотность костей будет снижаться. Такое состояние опасно частыми переломами.
В случае короткой длины влагалища для предотвращения диспареунии (боли во время полового акта) возможно хирургическое увеличения длины влагалища [20] .
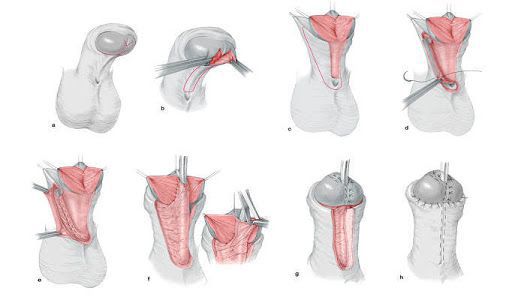
Больным с частичной андрогенной нечувствительностью с преимущественно мужским типом , которые выросли как мужчины, может быть предложено хирургическое лечение крипторхизма и гипоспадии. При крипторхизме проводится орхипексия — низведении неопущенного яичка в мошонку и фиксации его к окружающим тканям путём наложения шва. Коррекция гипоспадии подразумевает:
- восстановление отсутствующей части мочеиспускательного канала;
- восстановление нормального расположения мочеиспускательного канала;
- выпрямление полового члена;
- придание эстетически адекватного внешнего вида наружным половым органам.
В случае гинекомастии (увеличения груди) мужчинам может потребоваться маммопластика [21] . Часто пациентам с выявленным заболеванием требуется психологическая помощь.
Прогноз. Профилактика
Прогноз для жизни в случае синдрома Морриса благоприятный. Своевременная диагностика, гонадэктомия и заместительная гормональная терапия обычно дают хорошие краткосрочные и долгосрочные результаты. Пациентки часто адаптируются, нередко выходят замуж. Преодоление бесплодия возможно с помощью программ ЭКО с использованием донорских яйцеклеток, оплодотворённых спермой мужа, и суррогатного материнства. Главное — правильная адаптация и объяснение пациентке её состояния [12] .
Для профилактики синдрома тестикулярной феминизации женщинам, планирующим беременность рекомендуется пройти генетическое исследование, чтобы выяснить, не является ли она носителем патологического гена. Исследование должно проводится при отягощённом наследственном анамнезе, т. е. в том случае, если в семье были случаи андрогенной нечувствительности. Для профилактики остеопороза рекомендуется дополнительный приём кальция и витамина D [8] .
Синдром тестикулярной феминизации (Синдром Морриса) – наследственное заболевание, характеризующееся развитием женских половых признаков при наличии мужского кариотипа (XY). Симптомы этой патологии обладают широким спектром выраженности – от фенотипически полноценной женщины до полноценного мужчины с целым рядом промежуточных вариантов, на чем построена классификация данного состояния. Диагностика синдрома тестикулярной феминизации производится на основании результатов гинекологического или урологического осмотра, ультразвуковых исследований органов малого таза, изучения кариотипа и молекулярно-генетических анализов. Специфического лечения этого заболевания не существует, для улучшения качества жизни больных применяются разнообразные хирургические вмешательства.
Общие сведения
Синдром тестикулярной феминизации – генетическое заболевание, поражающее лиц с мужским кариотипом и приводящее к развитию у них разнообразных по выраженности женских половых признаков вплоть до полной феминизации. Нарушения подобного типа регистрировались давно, сразу после открытий хромосомных основ пола и исследований кариотипа, но данное заболевание впервые описал американских гинеколог Джон Моррис в 1953 году. Он изучил известных на тот момент (более 80) пациентов с мужским псевдогермафродитизмом и выявил ряд семейных случаев, что позволило ему определить синдром тестикулярной феминизации как X-сцепленное рецессивное наследственное заболевание.
В некоторых источниках эту патологию можно найти под названием синдрома Морриса. В настоящий момент установлено, что встречаемость синдрома тестикулярной феминизации составляет примерно 1 случай на 20-60 тысяч новорожденных мужского пола, однако частота носительства патологического гена среди женщин неизвестна. Данное заболевание является причиной почти 20% случаев мужского псевдогермафродитизма и обуславливает заметную долю от всех разновидностей первичной аменореи.
Причины
Исследования в области современной генетики позволили выявить молекулярно-генетические механизмы синдрома тестикулярной феминизации – таковыми оказались мутации гена AR, локализованного на X-хромосоме. Продуктом экспрессии данного гена является белок-рецептор к тестостерону и его метаболитам (в основном, дигидротестостерону), наличие которого и обеспечивает реакцию организма на мужские половые гормоны. На сегодняшний день выявлено более 300 различных типов мутаций гена AR, приводящих к синдрому тестикулярной феминизации. Все они имеют рецессивный характер, поэтому женщины (по кариотипу), имеющие гомологичную Х-хромосому, могут выступать только в качестве носителя и передавать это заболевание своим сыновьям с вероятностью 50%.
Из-за нарушений в структуре гена AR кодируемый им белок-рецептор получается дефектным, в зависимости от типа мутаций его реакция на воздействие тестостерона и сходных с ним соединений изменяется. При наиболее тяжелом течении синдрома тестикулярной феминизации рецептор становится совсем неспособным взаимодействовать с мужскими половыми гормонами, поэтому клетки организма теряют к ним чувствительность, сохраняя ее к эстрогенам (в основном, к эстрадиолу). Это приводит к развитию организма полностью по женскому типу при наличии и функционировании яичек. Некоторые другие типы синдрома тестикулярной феминизации обусловлены сохранением чувствительности к тестостерону, но на крайне низком уровне, что становится причиной широкого спектра клинических проявлений. Активность клеток Сертоли, выделяющих тестостерон, при любой форме синдрома тестикулярной феминизации сохраняется и может быть даже несколько повышена.
Классификация и симптомы
Неполная форма синдрома тестикулярной феминизации характеризуется намного более разнообразной клинической картиной. Как правило, причиной этой формы патологии выступают дефекты рецепторов к тестостерону, не приводящие к полной потере чувствительности, а только к ее значительному снижению или нарушению. Согласно классификации, принятой в 1996 году, выделяют пять основных форм или степеней неполного синдрома тестикулярной феминизации:
1. 1-я степень (мужской тип) характеризуется типично мужским фенотипом без каких-либо отклонений. В крайне редких случаях может наблюдаться высокий голос и признаки гинекомастии в подростковом возрасте. При этом практически всегда нарушены процессы сперматогенеза, поэтому у больных данным типом синдрома тестикулярной феминизации наблюдается мужское бесплодие.
2. 2-я степень (преимущественно мужской тип) – данный вариант заболевания проявляется более выраженными нарушениями вирилизации и формирования половых органов, хотя фенотипически больные являются мужчинами. У них часто обнаруживается гипоспадия, возможно развитие микропениса или сочетание этих признаков. Пациенты с этим вариантом синдрома тестикулярной феминизации нередко имеют гинекомастию и неравномерное отложение подкожной жировой клетчатки.
3. 3-я степень (амбивалентный тип) – характеризуется выраженным уменьшением полового члена, который становится похожим на клитор. Мошонка разделена настолько, что ее половины напоминают большие половые губы, часто наблюдается гипоспадия, крипторхизм. При этом типе синдрома тестикулярной феминизации также отмечается расширение таза, гинекомастия, относительно узкие плечи.
4. 4-я степень (преимущественно женский тип) – при этой форме синдрома тестикулярной феминизации больные являются женщинами по своим фенотипическим признакам, однако их клитор часто гипертрофирован, урогенитальный синус формирует короткое слепое влагалище. Нередко при этом варианте заболевания наблюдается такое нарушение, как сращение половых губ.
5. 5-я степень (женский тип) – больные этой формой синдрома тестикулярной феминизации фенотипически являются женщинами, практически никаких признаков вирилизации у них не обнаруживается за исключением несколько увеличенного размера клитора. В период полового созревания он может увеличиваться еще больше, достигая размеров микропениса.
У больных синдромом тестикулярной феминизации также часто возникают паховые грыжи из-за нарушения проходимости яичек по паховому каналу. Осложнениями гипоспадии могут быть разнообразные воспалительные процессы в мочевыделительной системе (уретриты, пиелонефриты). Крипторхизм грозит в будущем злокачественным перерождением тканей яичка, что является наиболее тяжелым осложнением данного заболевания.
Диагностика
Основными методами диагностики этого состояния являются гинекологический или урологический осмотр, ультразвуковое исследование, изучение наследственного анамнеза, молекулярно-генетический анализ и определение уровня половых гормонов. Раньше всего удается диагностировать неполные формы синдрома тестикулярной феминизации 2-5 степеней, так как нарушения в строении половых органов заметны уже при рождении ребенка. При осмотре врач-неонатолог может заподозрить наличие генетического заболевания и назначить дополнительные уточняющие исследования. Сочетание таких пороков развития с нормальным или даже повышенным уровнем тестостерона в крови и крипторхизмом говорит в пользу наличия синдрома тестикулярной феминизации.
Концентрация тестостерона при синдроме тестикулярной феминизации соответствует уровню здорового мужчины или даже несколько превышает его. При этом количество эстрогенов не достигает нижней отметки нормы для девушек аналогичного возраста. Изучение наследственного анамнеза может обнаружить признаки Х-сцепленной передачи заболевания. Молекулярно-генетическая диагностика синдрома тестикулярной феминизации производится врачом-генетиком при помощи автоматического секвенирования последовательности гена AR или других методик. Возможно также выявление носительства патологической формы гена у здоровых женщин и пренатальная диагностика этого заболевания.
Лечение синдрома тестикулярной феминизации
Лечение синдрома тестикулярной феминизации полного типа часто ограничивается простым удалением семенников с последующей коррекцией гормонального фона (при необходимости). Это требуется для профилактики семиномы и других форм злокачественного перерождения тканей яичка. Такие больные, с детства воспитывающиеся как девочки, после постановки диагноза могут нуждаться в психологической помощи. Терапия синдрома тестикулярной феминизации неполного типа характеризуется большим количеством пластических операций для воссоздания привычного вида половых органов, груди, оптимального функционирования мочевыделительной системы. Больные с любой формой этого заболевания бесплодны, что также зачастую требует помощи психологов.
Прогноз и профилактика
Прогноз синдрома тестикулярной феминизации относительно выживаемости больных довольно благоприятный – при полной форме патологии больные могут прожить нормальную жизнь женщины, обладая при этом мужским кариотипом. Неполные формы при правильно проведенной хирургической коррекции нарушений и пороков развития зачастую также не приводят к тяжелым и жизнеугрожающим осложнениям. Особенно высокую угрозу при синдроме тестикулярной феминизации представляет рак яичек в случае крипторхизма, поэтому его необходимо устранять – выводом семенников в мошонку (при мужском фенотипе) или удалением желез (в случае женского фенотипа).
Профилактика синдрома тестикулярной феминизации производится посредством генетического выявления носительства патологического гена при отягощенном наследственном анамнезе и в случае подтверждения – пренатальной диагностикой этой патологии.
Тестикулярная феминизация: причины и диагностика
Впервые тестикулярную феминизацию, или мужской гермафродитизм с общей феминизацией, описал Morris. Тестикулярная феминизация является типичной, хотя и редкой интерсексуальной формой первичной аменореи [Kaser]. Больные внешне имеют вид совершенно нормально развитых женщин с типично женским лицом, фигурой; психика без отклонений от нормы. Молочные железы, наружные половые органы развиты правильно, а оволосение, как правило, выражено слабо или совсем отсутствует (Уилкинс Л.).
Иногда оволосение бывает достаточно обильным [Teter et al.]. Влагалище может быть нормальным или уменьшенным в размере, но всегда оканчивается слепо. Иногда его совсем нет. Обычно матка и маточные трубы отсутствуют, иногда матка рудиментарна. Установлено семейное предрасположение к тестикулярной феминизации. Генетически больные имеют хроматиновый и гонадный мужской тип и в брюшной полости, а чаще в паховых грыжах (у большинства таких больных имеются двусторонние паховые грыжи) обнаруживаются семенники, изредка отмечаются гипертрофия клитора и нисхождение яичек вплоть до больших половых губ.
При гистологическом исследовании обнаруживаются различные формы дисгенетических яичек. Семенные канальцы могут быть хорошо развиты, а иногда не имеют просвета. Эпителий семенных канальцев состоит преимущественно из клеток типа Сертоли. Собственная оболочка некоторых семенных канальцев очень толстая и фиброзная. В таких канальцах клетки типа Сертоли, как правило, дегенерированы, клеток Лейдига немного, они небольшого размера. В некоторых случаях семенные канальцы очень малы, эпителий их недифференцирован. Интерстициальная ткань в небольшом количестве, фиброзно изменена и почти полностью лишена клеток Лейдига.
Характерно, что при тестикулярной феминизации клетки Лейдига, как правило, незрелы. Очень редко отмечаются случаи разрастания зрелых клеток Лейдига. Иногда описываются аденомы, состоящие из интерстициальных клеток, или новообразования с канальцевыми структурами типа adenoma tubulare [Morris, Teter, Boczowski].
Как полагают Kaser и Hammerstein, с патогенетической точки зрения данная форма гермафродитизма обусловлена первичной недостаточностью семенников или неправильной реакцией на воздействие андрогенов. Несмотря на нормальную закладку волосяных фолликулов, они оказываются резистентными к андрогенам, и даже вводимые извне андрогены не вызывают увеличения оволосения. Полагают, что резистентность к андрогенам проявляют все органы и системы организма.
Определение гормонов свидетельствует о нормальном или увеличенном количестве ФСГ, экскреции эстрогенов в пределах нормы для мужчин, а иногда выше нормы [Teter et al.]. 17-КС повышены до 20—30 мг или в норме, прегнандиол низкий, поливой хроматин отрицательный.
При отсутствии или слепо оканчивающемся влагалище, отсутствии матки при отрицательном половом хроматине и наличии семейной предрасположенности к тестикулярной феминизации диагноз очевиден. Интересно отметить, что Morris у 19 из 81 больной обнаружил тестикулярную феминизацию у сестер и теток. В сомнительных случаях показана лапаротомия с биопсией яичек.
Аплазия влагалища может встречаться у больных с женским генотипом, у которых нарушено развитие матки, влагалища в эмбриональном периоде, но с нормальными яичниками и маточными трубами (синдром Рокитанского — Кюстера). При лапароскопии диагноз уточняется.
Журнал: Проблемы репродукции. 2015;21(4): 43-47



Тестикулярная феминизация впервые описана Стегленером (G. Steglehner, 1817). В русской медицинской литературе эта патология впервые описана С.И. Благоволиным в 1893 г. Сам термин тестикулярная феминизация был введен Моррисом (J. Morris) в 1953 г. [4, 8]. Синдром тестикулярной феминизации (СТФ) (тестикулярная феминизация, синдром Морриса) обусловлен дефектом гена рецептора к андрогенам, который расположен на коротком плече Х-хромосомы. В процессе эмбриогенеза у этих больных гонады дифференцируются как яички, которые не способны к сперматогенезу, но секретируют тестостерон и вещество, ингибирующее мюллеровы протоки. Однако из-за дефекта гена андрогенных рецепторов отсутствует чувствительность к тестостерону, так как не продуцируется фермент 5-α редуктаза, превращающий тестостерон в биологически более активный дигидротестостерон, ответственный за формирование мужского фенотипа. Секреция эстрогенов надпочечниками и частично гонадами формирует женский фенотип при отсутствии производных мюллеровых протоков (маточных труб, матки и верхней трети влагалища) [1, 5, 7]. В результате этих процессов в ходе эмбрионального развития формируются наружные половые органы по нейтральному, женскому фенотипу: неполноценное укороченное влагалище, заканчивающееся слепо. Гонады (яички) расположены у стенок таза или (чаще) в паховых каналах или толще больших половых губ. Взрослые пациентки с полной формой СТФ [2, 6, 7], несмотря на кариотип 46XY и не опустившиеся яички (гонады), отличаются женским телосложением с развитыми молочными железами, скудным лобковым и подмышечным оволосением, отсутствием внутренних половых органов и слепо заканчивающимся влагалищным мешком. Глубина влагалища может варьировать от нормальной до укороченной.
СТФ относится к редким формам генетических заболеваний. Комплексное обследование таких больных состоит из медико-генетического консультирования, кариотипирования, определения половых гормонов в сыворотке крови, ультрасонографии органов малого таза.
Согласно современным представлениям, гонады у пациентов с интерсексуальными состояниями обладают высоким риском малигнизации. Гонадобластома тестикул встречается в 20—50% наблюдений. В связи с этим половые железы рекомендуют сохранять до завершения пубертатного периода и конституционального формирования, а затем проводить удаление гонад. Хирургическая тактика в отношении тестикул в настоящее время сводится к лапароскопической гонадэктомии [3, 6]. Заместительная гормональная терапия эстрогенами достаточна для дальнейшего формирования женского фенотипа и профилактики остеопороза.
Представленный случай СТФ прослежен с 1997 г. от момента диагностики и проведения оперативного лечения (гонадэктомии) методом оперативной лапароскопии до 2014 г.
Пациентка К., 14 лет, впервые в 1997 г. обратилась в Межрегиональный центр планирования семьи и репродукции человека (отделение Центра Охраны материнства и детства, Симферополь, Крым) с жалобами на первичную аменорею. Из анамнеза: в четырехлетнем возрасте проведена правосторонняя паховая герниопластика. Детскими хирургами в составе грыжевого содержимого обнаружена ткань, напоминающая по структуре яичко. Генетическое обследование пациентки установило кариотип 46XY. К врачам родители не обращались до 14 лет ребенка. При осмотре — фенотип женский, телосложение нормостеническое, рост — 170 см, вес — 50 кг, молочные железы развиты соответственно возрасту. Интеллект сохранен. Самоосознание, поло-ролевое поведение и психосоциальная ориентация женские. Подмышечное и лобковое оволосение отсутствует. Наружные половые органы полностью соответствуют женскому фенотипу, девственная плева отсутствует. Зондирование влагалища (длина по зонду 7 см), осмотр с помощью детского зеркала Куско № 1: влагалище узкое, заканчивается слепо, шейка матки не визуализируется. Ректальный осмотр выявил отсутствие матки и придатков. При ультразвуковом исследовании в малом тазу в области правой гонады визуализируется гипоэхогенное образование 25×17 мм, с анэхогенным включением 21 мм, в области проекции левой гонады — гиперэхогенное образование 21×18 мм (рис. 1).

Рис. 1. Ультразвуковое исследование в области правой гонады при первичном осмотре пациентки К., 1997 г.
Гормональное обследование методом ИФА: тестостерон — 26,21 нмоль/л (норма для женщин старше 10 лет — 0,45—3,75 нмоль/л, у мужчин старше 14 лет — 5,76 — 28,14 нмоль/л); пролактин — 107 МЕ/л.
На основании клинических данных, результатов УЗИ, исследования кариотипа и лабораторных показателей был поставлен диагноз: полная форма синдрома тестикулярной феминизации (СТФ).
Учитывая риск малигнизации гонад в постпубертатном периоде у пациентов с СТФ рекомендована гонадэктомия.
После беседы с родителями и получения их согласия, девочке проведена оперативная лапароскопия (1997 г.). В ходе операции при ревизии малого таза подтверждено отсутствие матки, маточных труб, связок и яичников с двух сторон. Слева, в области проекции паховой связки, экстраперитонеально визуализировалось опухолевидное образование размером 30×25×25 мм, мягкой консистенции, с тяжем, идущим к стенкам таза. Справа, в области проекции внутреннего кольца пахового канала — расположенное экстраперитонеально опухолевидное образование 25×25×20 мм, подпаяно к передней брюшной стенке, мягковатой консистенции. Гистологическое исследование по cito полученных тканей: слева — в состав фиброзных оболочек включено дизонтогенетически незрелое образование, морфологически без признаков малигнизации. Участок незрелой ткани яичка, cправа — многокомпонентная тератома яичка.
Произведено удаление образований путем наложения с обеих сторон по три петли Редера с последующим отсечением и коагуляцией культей. Окончательное гистологическое заключение: левая гонада — дисплазия дистопированных яичек в сочетании; правая гонада — участок фиброзно-мышечной ткани с кистозно-расширенным протоком, жировой клетчатки по периферии (возможно, во время герниопластики в детстве правое яичко было удалено или частично удалено).
Послеоперационный период протекал без осложнений.
В качестве заместительной гормональной терапии (ЗГТ) после операции назначен эстриол (Овестин по 2 мг в день).
Динамическое наблюдение за пациенткой после операции:
8 месяцев после операции. Принимает в непрерывном режиме КОК (Марвелон по 1 таблетке 1 раз в сутки). На фоне терапии уровни ЛГ, ФСГ в пределах нормы; тестостерон 3,9 нмоль/л (норма у женщин 0,45—3,75 нмоль/л). При проведении сонографии органов малого таза патологических образований не выявлено.
3 года после операции. В качестве ЗГТ принимает эстрадиол (Дивигель 0,5 г 1 раз в 3 дня). Волосяной покров в подмышечных впадинах и на лобке отсутствует. Тестостерон — 29,7 нмоль/л (норма у женщин — 0,45—3,75 нмоль/л). При проведении сонографии органов малого таза патологических образований не выявлено.
Пациентка переезжает в Москву, к врачам не обращается, приезжая к родителям в Крым иногда приходит на прием.
5 лет после операции. Принимает 17-β эстрадиол (Эстрожель 1,25 г геля 1 раз в 3 дня). При проведении сонографии органов малого таза патологических образований не выявлено. Гормональная диагностика не проводилась. Молочные железы развиты за счет жировой ткани (рис. 2).

Рис. 2. Эхографическая картина молочных желез у пациентки К., 2004 г.
В 2004 г. пациентка выходит замуж.
23 декабря 2008 г. проведена вторая лапароскопия в Москве: справа забрюшинно, тотчас ниже внутреннего отверстия пахового канала определяется образование размером 25×35 мм железистой структуры. Выполнено удаление образования справа. Окончательное гистологическое заключение: ткань яичка. Послеоперационное течение без осложнений. Выписана через сутки после операции.
1 год и 3 мес после повторной операции (2010 г.). Получает 17-β эстрадиол (Эстрожель 1,25 г 1 раз в 3 дня). Тестостерон 0,55 нмоль/л. УЗИ — в малом тазу патологических образований не выявлено (рис. 6).
Живет и активно работает на престижной работе. Второй раз вышла замуж. Хочет создать полноценную семью.
В 2013 г. проведена программа ВРТ — донорство ооцитов, сперма мужа, суррогатное материнство. Наступила беременность двойней. Стала мамой двоих детей.
Данные динамического наблюдения показывали рост концентрации тестостерона уже в первые годы после первой операции, что свидетельствовало о функционировании не полностью удаленной гонады в 1997 г. (очаг гиперпродукции андрогенов), и что подтвердилось через 8 лет при ультразвуковом контроле.
Выражаю признательность врачу — хирургу Панину Александру Викторовичу (в 2008 г. зав. эндоскопическим отделением ЦПСиР, Москва), который провел пациентке повторное оперативное вмешательство.
Читайте также: