
Доклад на тему адреногенитальный синдром
Обновлено: 08.07.2024
Какая связь между недостатком в организме гормона, контролирующего обмен углеводов и опосредующего стресс-реакции, и множественными нарушениями полового развития? Самая прямая. Примером служит т.н. адреногенитальный синдром у детей и взрослых. Этот довольно редкий наследственный недуг поражает людей обоего пола (в среднем — одного на пять–шесть тысяч) и уже в юном возрасте имеет свои характерные проявления.
В результате, желая скомпенсировать эти недостатки, организм запускает каскад взаимодействий между различными органами, имеющий патологические следствия. Если более конкретно:
- нехватка гормонов провоцирует мозговые структуры (точнее, гипоталамо-гипофизарную систему) к выработке фактора, побуждающего кору надпочечников наверстать упущенное;
- в ответ на этот однозначный стимул надпочечники силятся исполнить приказ и увеличивают количество рабочих клеток (т.н. гиперплазия);
- но, поскольку необходимые ферменты по-прежнему отсутствуют, образование кортизола (и, в некоторых случаях, альдостерона) не приходит в норму;
- зато без всякой необходимости (на фоне исходно нормальной выработки половых стероидов) усиливается продукция все той же корой надпочечников веществ, обладающих потенциалов половых гормонов — андрогенов (еще до рождения!);
- избыток половых гормонов производит в организме всевозможные эффекты;
- в то время как недостаток кортизола (и, в некоторых случаях, альдостерона) по-прежнему дает о себе знать и провоцирует мозг на дальнейшую стимуляцию работы коры надпочечников.
Две другие формы (редкие) — гипертоническая и сольтеряющая — более тяжелые. Настолько, что без их раннего выявления существует высокий риск гибели детей от сердечной и почечной недостаточности, и кровоизлияния в мозг.
Симптомы адреногенитального синдрома
Адреногенитальный синдром у новорожденных
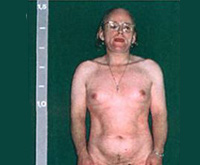
Поскольку надпочечники под руководством мозговых центров (в ответ на нехватку кортизола) начинают усиленно трудиться еще внутриутробно, ребенок рождается на свет, уже имея избыток половых гормонов. В результате у новорожденных девочек регистрируют симптомы т.н. ложного гермафродитизма (наличия признаков обоих полов). А у новорожденных мальчиков — увеличенный копулятивный орган. Кроме того, аномальное количество половых стероидов приводит к избыточной пигментации наружных половых органов, кожи вокруг ануса и сосков. Перечисленные признаки характерны для самой распространенной (вирильной) формы адреногенитального синдрома.
При более редкой и тяжелой форме (сольтеряющей) к ним добавляются следствия потери организмом натрия и хлора, и задержки калия (из-за недостатка альдостерона):
- диспепсические расстройства (рвота, не связанная с кормлением), понос;
- обезвоживание (в том числе, вследствие потери жидкости с рвотой и жидким стулом);
- судороги ;
- низкое артериальное давление;
- нарушения ритма сердца.
Адреногенитальный синдром у девочек и мальчиков
- увеличение половых органов;
- оволосение подмышек и лобковой области;
- изменение тембра голоса;
- появление угревой сыпи;
- высокие темпы роста и набора массы тела и др.
Касательно последнего пункта: высокие темпы роста характерны только для юного возраста. Довольно рано (к двенадцати годам) зоны роста закрываются, рост сильно притормаживается или останавливается вовсе. В результате в подростковом периоде наблюдается уже отставание в росте от сверстников.
Другими (неспецифическими) признаками не выявленных в очень раннем детстве гипертонической и сольтеряющей форм адреногенитального синдрома могут стать:
- стойкое повышение артериального давления;
- диспепсические нарушения.
Длительная гипертензия рискует осложниться ухудшением зрения, сердечно-сосудистыми, почечными и мозговыми нарушениями (вплоть до инсульта).
Для правильной (и ранней) диагностики помимо осмотра и опроса необходимы лабораторные и инструментальные исследования:
- анализы крови и мочи на гормоны и их предшественники;
- ультразвуковое исследование;
- рентгенография кистей рук;
- магниторезонансная и компьютерная томография.
Лечение адреногенитального синдрома
Чем раньше поставлен правильный диагноз, тем эффективнее лечение.
Прежде всего, оно состоит в заместительной терапии недостающими гормонами (на протяжении всей жизни человека). Хорошим подспорьем медикаментозным методам служат: диета с учетом формы патологии и индивидуальными особенностями организма и психотерапевтические сеансы. Возможно хирургическое вмешательство — удаление клитора, пластика влагалища.
Адреногенитальный синдром — наследственное заболевание надпочечников, при котором вследствие функциональной несостоятельности ферментов нарушается стероидогенез. Проявляется вирилизацией гениталий, маскулиноподобным телосложением, недоразвитием груди, гирсутизмом, акне, аменореей или олигоменореей, бесплодием. В ходе диагностики определяют уровни 17-гидроксипрогестерона, 17-кетостероидов, андростендиона, АКТГ, проводят УЗИ яичников. Пациенткам назначают заместительную гормонотерапию глюкокортикоидами и минералокортикоидами, эстрогены в комбинации с андрогенами или прогестинами нового поколения. При необходимости выполняют пластику половых органов.
МКБ-10
Общие сведения
Адреногенитальный синдром, или врожденная дисфункция (гиперплазия) коры надпочечников, — наиболее частое из наследуемых заболеваний. Распространенность патологии отличается у представителей разных национальностей. Классические варианты АГС у лиц европеоидной расы встречаются с частотой 1:14 000 младенцев, в то время как у эскимосов Аляски этот показатель составляет 1:282.
Существенно выше заболеваемость у евреев. Так, неклассическую форму адреногенитального расстройства выявляют у 19% лиц еврейской национальности группы ашкенази. Патология передается по аутосомно-рецессивному типу. Вероятность рождения ребенка с таким синдромом при носительстве патологического гена у обоих родителей достигает 25%, в браке носителя и больного — 75%. Если один из родителей имеет полноценные ДНК, клинические проявления синдрома у детей не развиваются. При наличии АДС у отца и матери ребенок также будет болен.
Причины
У больных с наследуемой гиперплазией надпочечников генетический дефект проявляется несостоятельностью ферментных систем, участвующих в секреции стероидных гормонов. В 90-95% случаев патология возникает при повреждении гена, который отвечает за синтез 21-гидроксилазы — фермента, влияющего на образование кортизола. В остальных клинических случаях вследствие дефекта ДНК нарушается производство других ферментов, обеспечивающих стероидогенез, — StAR/20,22-десмолазы, 3-β-гидрокси-стероиддегидрогеназы, 17-α-гидроксилазы/17,20-лиазы, 11-β-гидроксилазы, P450-оксидоредуктазы и синтетазы альдостерона.
У пациентов с признаками вирилизирующего синдрома вместо активного гена CYP21-B в коротком плече 6-й аутосомы расположен функционально несостоятельный псевдоген CYP21-A. Структура этих участков ДНК-цепи во многом гомологична, что повышает вероятность конверсии генов в мейозе с перемещением участка нормального гена на псевдоген или делецию CYP21-B.
По-видимому, именно этими механизмами объясняется существование скрытых форм болезни, дебютирующих в пубертате или постпубертатном периоде. В таких случаях клинические признаки патологии становятся заметными после нагрузок, истощающих кору надпочечников: тяжелых болезней, травм, отравлений, радиационных воздействий, длительного периода интенсивной работы, психологически напряженных ситуаций и т. д.
Патогенез
В основе механизма развития наиболее распространенного варианта адреногенитального синдрома с дефектом CYP21-B-гена лежит принцип обратной связи. Ее начальным звеном становится дефицит стероидов — кортизола и альдостерона. Несостоятельность процессов гидроксилирования сопровождается неполным переходом 17-гидроксипрогестерона и прогестерона в 11-дезоксикортизол и дезоксикортикостерон. В результате снижается секреция кортизола, а для компенсации этого процесса в гипофизе усиливается синтез АКТГ — гормона, вызывающего компенсаторную гиперплазию коры надпочечников для стимуляции выработки кортикостероидов.
Параллельно возрастает синтез андрогенов и появляются видимые признаки их влияния на чувствительные ткани и органы. При умеренном снижении активности фермента минералокортикоидная недостаточность не развивается, поскольку потребность организма в альдостероне почти в 200 раз ниже по сравнению с кортизолом. Только глубокий дефект гена вызывает тяжелую клиническую симптоматику, которая проявляется с раннего возраста. Патогенез развития заболевания при нарушении структуры других участков ДНК аналогичен, однако пусковым моментом являются нарушения в других звеньях стероидогенеза.
Классификация
Систематизация различных форм вирилизирующей гиперплазии надпочечников основана на особенностях клинической картины заболевания, выраженности генетического дефекта и времени проявления первых патологических признаков. Тяжесть расстройства напрямую связана со степенью повреждения ДНК. Специалисты в сфере эндокринологии различают следующие виды адреногенитального синдрома:
- Сольтеряющий. Самый тяжелый вариант патологии, проявляющийся в первый год жизни ребенка грубыми нарушениями строения наружных половых органов у девочек и их увеличением у мальчиков. Активность 21-гидроксилазы составляет не более 1%. Значительное нарушение стероидогенеза приводит к выраженным соматическим нарушениям — рвоте, поносу, судорогам, чрезмерной пигментации кожи. Без лечения такие дети умирают в раннем возрасте.
- Простой вирильный. Течение заболевания менее тяжелое, чем при сольтеряющем варианте. Преобладают проявления неправильного развития гениталий у младенцев женского пола, увеличение их размеров у мальчиков. Признаки надпочечниковой недостаточности отсутствуют. Уровень активности 21-гидроксилазы снижен до 1-5%. С возрастом у пациентов нарастают признаки вирилизации вследствие стимулирующего действия андрогенов.
- Неклассический (постпубертатный). Наиболее благоприятная форма АГС, явные признаки которой возникают в период полового созревания и в репродуктивном возрасте. Наружные половые органы имеют нормальное строение, может быть увеличен клитор у женщин и половой член у мужчин. Функциональность 21-гидроксилазы снижена до 20-30%. Заболевание выявляется случайно при обследовании в связи с бесплодием или нарушениями менструальной функции.
Сольтеряющий и простой вирильный виды адреногенитальных расстройств относят к категории антенатальной патологии, формирующейся внутриутробно и проявляющейся с момента рождения. При дефекте строения других генов наблюдаются более редкие варианты заболевания: гипертензивные — классический (врожденный) и неклассический (поздний), гипертермический, липидный, с ведущими проявлениями гирсутизма.
Симптомы
Сольтеряющий и простой вирильный
При антенатальных формах заболевания основным клиническим симптомом является видимая вирилизация гениталий. У новорожденных девочек обнаруживаются признаки женского псевдогермафродитизма. Клитор большой по размерам или имеет пенисообразную форму, преддверие влагалища углублено, сформирован урогенитальный синус, большие и малые половые губы увеличены, промежность высокая. Внутренние половые органы развиты нормально.
У младенцев-мальчиков увеличен половой член и гиперпигментирована мошонка. Кроме того, при сольтеряющем адреногенитальном расстройстве выражена симптоматика надпочечниковой недостаточности с тяжелыми, зачастую несовместимыми с жизнью соматическими нарушениями (понос, рвота, судороги, обезвоживание и др.), которые проявляются с 2-3-недельного возраста. У девочек с простым вирильным АГС по мере взросления признаки вирилизации усиливаются, формируется диспластическое телосложение.
Из-за ускорения процессов окостенения пациентки отличаются невысоким ростом, широкими плечами, узким тазом, короткими конечностями. Трубчатые кости массивные. Половое созревание начинается рано (до 7 лет) и протекает с развитием вторичных мужских половых признаков. Отмечается увеличение клитора, снижение тембра голоса, нарастание мышечной силы, формирование типичной для мужчин формы перстневидного хряща щитовидной железы. Грудь не растет, менархе отсутствует.
Неклассический
Менее специфичны клинические симптомы при неклассических формах вирилизирующего синдрома, возникшие в пубертате и после стрессовых нагрузок (выкидыша на ранних сроках беременности, медицинского аборта, операции и др.). Обычно пациентки вспоминают, что у них еще в младшем школьном возрасте появилось небольшое оволосение в подмышечных впадинах и на лобке. В последующем развились признаки гирсутизма с ростом стержневых волос над верхней губой, по белой линии живота, в области грудины, в сосково-ареолярной зоне.
Женщины с АГС предъявляют жалобы на стойкую угревую сыпь, пористость и повышенную жирность кожи. Менархе наступает поздно — к 15-16 годам. Менструальный цикл неустойчив, интервалы между менструациями достигают 35-45 дней и более. Кровянистые выделения во время месячных скудные. Молочные железы небольшие. Клитор несколько увеличен. Такие девушки и женщины могут иметь высокий рост, узкий таз, широкие плечи.
По наблюдениям специалистов в сфере акушерства и гинекологии, чем позже развиваются адреногенитальные расстройства, тем менее заметны внешние признаки, характерные для мужчин, и тем чаще ведущим симптомом становится нарушение месячного цикла. При более редких генетических дефектах пациентки могут жаловаться на повышение артериального давления или, наоборот, гипотонию с низкой работоспособностью и частыми головными болями, гиперпигментацию кожи с минимальными симптомами вирилизации.
Осложнения
Основным осложнением адреногенитального синдрома, по поводу которого пациентки обращаются к акушерам-гинекологам, является стойкое бесплодие. Чем раньше проявилось заболевание, тем меньше вероятность забеременеть. При значительной ферментной недостаточности и клинических проявлениях простого вирилизирующего синдрома беременность вообще не наступает.
У забеременевших пациенток с пубертатными и постпубертатными формами заболевания возникают самопроизвольные выкидыши на раннем сроке. В родах возможна функциональная истмико-цервикальная недостаточность. Такие женщины более склонны к возникновению психоэмоциональных расстройств — склонности к депрессии, суицидальному поведению, проявлениям агрессии.
Диагностика
Постановка диагноза при антенатальных типах АГС с характерными изменениями половых органов не представляет сложности и проводится сразу после родов. В сомнительных случаях применяют кариотипирование для подтверждения женского кариотипа (46ХХ), молекулярно-генетические тесты. Большее значение диагностический поиск приобретает при позднем клиническом дебюте или скрытом течении с минимальными внешними проявлениями вирилизации. В подобных ситуациях для выявления адреногенитального синдрома используют следующие лабораторные и инструментальные методы:
- Уровень 17-ОН-прогестерона. Высокая концентрация 17-гидроксипрогестерона, который является предшественником кортизола — ключевой признак недостаточности 21-гидроксилазы. Его содержание увеличено в 3-9 раз (от 15 нмоль/л и выше).
- Стероидный профиль (17-КС). Повышение уровня 17-кетостероидов в моче у женщин в 6-8 раз свидетельствует о высоком содержании андрогенов, производимых корой надпочечников. При выполнении преднизолоновой пробы концентрация 17-КС уменьшается на 50-75%.
- Содержание андростендиона в сыворотке крови. Повышенные показатели этого высокоспецифичного метода лабораторной диагностики подтверждают усиленную секрецию предшественников мужских половых гормонов.
- Уровень АКТГ в крови. Для классических форм заболевания характерна компенсаторная гиперсекреция адренокортикотропного гормона передней долей гипофиза. Поэтому при синдроме вирилизирующей дисфункции показатель повышен.
- УЗИ яичников. В корковом веществе определяются фолликулы на разных стадиях созревания, не достигающие преовуляторных размеров. Яичники могут быть несколько увеличены, однако разрастания стромы не наблюдается.
- Измерение базальной температуры. Температурная кривая типична для ановуляторного цикла: первая фаза растянута, вторая укорочена, что обусловлено недостаточностью желтого тела, которое не образуется из-за отсутствия овуляции.
Для сольтеряющего варианта АГС также характерна повышенная концентрация ренина в плазме крови. Дифференциальная диагностика адреногенитальных расстройств, возникших в пубертатном и детородном возрасте, проводится с синдромом поликистозных яичников, овариальными андробластомами, андростеромами надпочечников, вирильным синдромом гипоталамического происхождения и конституциональным гирсутизмом. В сложных случаях к диагностике привлекают эндокринологов, урологов, врачей-генетиков.
Лечение адреногенитального синдрома
Основным способом коррекции вирильной дисфункции надпочечников является заместительная гормональная терапия, восполняющая дефицит глюкокортикоидов. Если у женщины со скрытым АГС нет репродуктивных планов, кожные проявления гиперандрогении незначительны и месячные ритмичны, гормоны не применяют. В остальных случаях выбор схемы лечения зависит от формы эндокринной патологии, ведущей симптоматики и степени ее выраженности. Зачастую назначение глюкокортикоидных препаратов дополняют другими медикаментозными и хирургическими методами, подобранными в соответствии с конкретной терапевтической целью:
- Лечение бесплодия. При наличии планов по деторождению женщина под контролем андрогенов крови принимает глюкокортикоиды до полного восстановления овуляторного месячного цикла и наступления беременности. В резистентных случаях дополнительно назначают стимуляторы овуляции. Во избежание выкидыша гормонотерапию продолжают до 13-й недели гестационного срока. В I триместре также рекомендованы эстрогены, во II-III — аналоги прогестерона, не обладающие андрогенным эффектом.
- Коррекция нерегулярных месячных и вирилизации. Если пациентка не планирует беременность, но жалуется на расстройство менструального цикла, гирсутизм, угри, предпочтительны средства с эстрогенным и антиандрогенным эффектом, оральные контрацептивы, содержащие гестагены последнего поколения. Терапевтический эффект достигается за 3-6 месяцев, однако по окончании лечения при отсутствии заместительной гормонотерапии признаки гиперандрогении восстанавливаются.
- Лечение врожденных форм АГС. Девочкам с признаками ложного гермафродитизма проводят адекватную гормонотерапию и выполняют хирургическую коррекцию формы половых органов — клитеротомию, интроитопластику (вскрытие урогенитального синуса). При сольтеряющих адреногенитальных расстройствах кроме глюкокортикоидов под контролем рениновой активности назначают минералокортикоиды с увеличением терапевтических доз при возникновении интеркуррентных заболеваний.
Определенные сложности в ведении пациентки возникают в тех случаях, когда заболевание не диагностировано в акушерском стационаре, и девочка с выраженной вирилизацией гениталий регистрируется и воспитывается как мальчик. При решении о восстановлении женской половой идентичности хирургическую пластику и гормонотерапию дополняют психотерапевтической поддержкой. Решение о сохранении гражданского мужского пола и удалении матки с придатками принимается в исключительных случаях по настоянию больных, однако такой подход считается ошибочным.
Прогноз и профилактика
Прогноз при своевременном обнаружении адреногенитального синдрома и адекватно подобранной терапии благоприятный. Даже у пациенток со значительной вирилизацией гениталий после пластической операции возможна нормальная половая жизнь и естественные роды. Заместительная гормонотерапия при любой форме АГС способствует быстрой феминизации — развитию грудных желез, появлению месячных, нормализации овариального цикла, восстановлению генеративной функции. Профилактика заболевания осуществляется на этапе планирования беременности.
Если в роду наблюдались случаи подобной патологии, показана консультация генетика. Проведение пробы с АКТГ обоим супругам позволяет диагностировать гетерозиготное носительство или скрытые формы адреногенитального расстройства. При беременности синдром может быть обнаружен по результатам генетического анализа клеток хорионических ворсин или содержимого околоплодных вод, полученных методом амниоцентеза. Неонатальный скрининг, проводимый на 5-е сутки после родов, направлен на выявление повышенной концентрации 17-гидропрогестерона для быстрого выбора терапевтической тактики.

Надпочечники выделяют в кровь множество гормонов, регулирующих рост и развитие организма. Адреногенитальный синдром – наследственное заболевание, характеризующееся нарушением этой функции. При классическом течении болезни избыточный синтез андрогенов в надпочечниках начинается еще на 9 – 10 неделе беременности, что приводит к неправильному формированию половых органов.
Адреногенитальный синдром и его формы
В 90% случаев развитие болезни связано с отсутствием фермента гидроксилазы. Это ведет к тяжелым нарушениям синтеза гормонов. Врожденный адреногенитальный синдром встречается у 1 из 5000 новорожденных. Заболевание сопровождается усилением активности коры надпочечников и увеличением концентрации в крови мужских половых гормонов – андрогенов.
Кроме ферментативных нарушений, в развитии патологии может иметь значение опухоль надпочечников. Она ведет к развитию приобретенного варианта заболевания.
Формы адреногенитального синдрома:

В классификации также представлены более редкие гипертензивная форма с задержкой в организме воды и солей и развитием стойкой гипертензии, липидная и гипертермическая формы.
Причины
Причины адреногенитального синдрома связаны с генетическим нарушением – дефектом одного из генов 6-1 пары хромосом. Этот ген рецессивный, и проявляется только в случае рождения ребенка с обеими измененными хромосомами. Его родители обязательно должны быть либо носителями патологического гена, либо страдать этим заболеванием.
Этот ген отвечает за синтез фермента 21-гидроксилазы. Ее недостаток или отсутствие приводят к компенсаторному усиленному синтезу гормона гипофиза АКТГ, что ведет к гипертрофии надпочечников и к усилению выработки андрогенов. Они подавляют функцию яичников и образование женских половых гормонов – эстрогенов.
Почему происходит мутация этого гена, неизвестно. Носителями измененного гена с равной частотой могут быть и мальчики, и девочки. Клиническое развитие заболевания тоже не зависит ни от пола ребенка, ни от того, кто из родителей болен, а кто лишь передал патологический ген.
Симптомы и диагностика заболевания

Больные должны наблюдаться у детского эндокринолога. Грамотное лечение именно у этого специалиста дает возможность нормализовать гормональную функцию.
Симптомы адреногенитального синдрома:
- быстрый рост в подростковом возрасте;
- возникновение у девушек явлений гирсутизма – волос на верхней губе, внутренней поверхности бедер, по белой линии живота, вокруг сосков;
- выраженная угревая сыпь;
- нарушения менструального цикла, бесплодие.
Внешние признаки адреногенитального синдрома наиболее заметны у девушек:
- мужской тип телосложения – высокий рост, узкий таз, недоразвитые молочные железы, широкие плечи; вес в пределах нормы;
- нарушение строения наружных половых органов при нормально сформированной матке и естественной для женщины сексуальной ориентации;
- при наступлении беременности частое осложнение – прерывание в 1-м триместре.
Диагностика адреногенитального синдрома проводится с помощью таких исследований:
- определение уровня гормонов: тестостерона, ДЭА, ДЭА-С, 17-ОНП, ЛГ, ФСГ, показывающих степень нарушения функций надпочечников и яичников;
- пробы с дексаметазоном и АКТГ, необходимые для дифференциальной диагностики с синдромом поликистозных яичников; яичников;
- определение длительности фаз цикла путем составления графика базальной температуры.
Лечение
При сольтеряющей форме заболевания лечение начинают сразу после рождения ребенка, как только появились первые симптомы. Оно включает интенсивную терапию с введением жидкости, солей, гормональных препаратов.
Лечение адреногенитального синдрома включает:
- длительный прием гормонов надпочечников – глюкокортикоидов, который должен начинаться в раннем детстве (до 7 лет);
- по показаниям – назначение женских половых гормонов, особенно при подготовке к беременности;
- при необходимости еще в детском возрасте пациенткам проводят хирургическую пластику наружных половых органов.
Значительную трудность вызывает сохранение беременности у больных женщин. Однако грамотный гинеколог-эндокринолог способен подобрать необходимые лекарственные препараты, чтобы пациентка смогла родить здорового ребенка.
Профилактика
Единственный способ профилактики адреногенитального синдрома – генетическая консультация будущих родителей с определением у них мутированного гена. Если хотя бы у одного из родителей такой мутации нет, то у пары родится клинически здоровый ребенок.
- лабораторная диагностика гормональных нарушений на современном оборудовании;
- прием профильных специалистов – педиатра, гинеколога, эндокринолога;
- последовательное наблюдение за ребенком, преемственность в лечении у докторов разного профиля;
- современные методы лечения при вынашивании беременности;
- назначение лекарственной терапии по современным методикам.
Врачи нашей клиники проводят раннюю диагностику и назначают грамотное лечение при любой форме заболевания. В таких условиях проявления адреногенитального синдрома хорошо корректируются. Записывайтесь на прием прямо сейчас!
Отзывы
Хорошая клиника, хороший врач! Раиса Васильевна может понятно и доступно объяснить, в чем суть проблемы. Если что-то не так, она обо всем говорить прямо, не завуалированно, как это порой делают другие врачи. Не жалею, что попала именно к ней.
Хочу выразить благодарность работникам клиники Мама, Папа, я. В клинике очень дружелюбная атмосфера, очень приветливый и веселый коллектив и высококвалифицированные специалисты. Спасибо вам большое! Желаю процветания вашей клинике.
Сегодня удаляла родинку на лице у дерматолога Кодаревой И.А. Доктор очень аккуратная! Корректная! Спасибо большое! Администратор Борщевская Юлия доброжелательна, четко выполняет свои обязанности.
Сегодня обслуживалась в клинике, осталась довольна персоналом, а также врачом гинекологом. Все относятся к пациентам с уважением и вниманием. Спасибо им большое и дальнейшего процветания.
Первое посещение понравилось. Меня внимательно осмотрели, назначили дополнительные обследования, дали хорошие рекомендации. Буду продолжать лечение и дальше, условия в клинике мне понравились.
Доктор внимательно осмотрела моего мужа, назначила ЭКГ и поставили предварительный диагноз. Дала рекомендации по нашей ситуации и назначила дополнительное обследование. Пока замечаний нет. Финансовые договоренности соблюдены.
Очень понравилась клиника. Обходительный персонал. Была на приеме у гинеколога Михайловой Е.А. осталась довольна, побольше таких врачей. Спасибо.

Адреногенитальный синдром — это генетическое заболевание с аутосомно-рецессивным типом наследования, в основе которого лежит дефект одного из ферментов или транспортных белков, принимающих участие в биосинтезе кортизола в коре надпочечников. На сегодняшний день известно 7 форм адреногенитального синдрома (АГС), или врожденной дисфункции коры надпочечников (ВДКН), в зависимости от типа ферментативного дефицита:
- Дефект STAR;
- Дефицит 20,22-десмолазы (11α-гидроксилазы);
- Дефицит 17α-гидроксилазы/17,20-лиазы;
- Дефицит 3β-гидроксистероиддегидрогеназы;
- Дефицит 21-гидроксилазы;
- Дефицит 11β-гидроксилазы;
- Дефицит оксидоредуктазы.
Инициирующим звеном в патогенезе адреногенитального синдрома являются мутации гена, кодирующего стероид-21-гидроксилазу (CYP21, CYP21B, CYP21A2), и гомологичного ему псевдогена (CYP21P, CYP21A, CYP21A1P), которые локализованы в HLA-комплексе между локусами HLA-B и HLA-DR на коротком плече 6-й хромосомы (6р21.3). За счет особенностей организации высокогомологичных генов это приводит к частым рекомбинациям между ними. Большинство описанных мутаций (90–95 %) в свою очередь ведут к 21-гидроксилазной недостаточности и являются результатом двух типов рекомбинаций между геном CYP21 и псевдогеном CYP21P. Первый механизм — неравный кроссинговер во время мейоза, приводящий к частичной делеции гена CYP21 и замещению большого фрагмента гена CYP21 аналогичным фрагментом псевдогена CYP21P. Второй механизм — генная конверсия, включающая в себя точечные мутации, обычно присутствующие в псевдогене CYP21P, в активный ген CYP21.
Недостаточность фермента 21-гидроксилазы вызывает нарушение синтеза кортизола, что по механизму отрицательной обратной связи стимулирует корковый слой надпочечников и приводит к его гиперплазии. В то же время данный ферментативный блок служит причиной чрезмерного накопления предшественников кортизола и андрогенов. Схема патогенеза ВДКН представлена ниже.
В клинической картине адреногенитального синдрома выделяют два ведущих симптома. Это, во-первых, надпочечниковая недостаточность из-за сниженного синтеза кортизола и альдостерона, а во-вторых — гиперандрогения из-за повышенной продукции половых стероидов. На основании уровня активности фермента 21-гидроксилазы выделяют две классические формы АГС: сольтеряющую и вирильную.
Для сольтеряющей формы АГС характерен дефицит как минералокортикоидов, так и глюкокортикоидов. При этом отсутствие компенсаторных реакций может привести к сольтеряющему кризу, обусловленному снижением реабсорбции натрия в канальцах почки, а также снижением ОЦК и АД, что приводит к резко выраженному обезвоживанию уже в первые дни после рождения.
При вирильной форме АГС снижается только синтез кортизола, что проявляется мышечной слабостью, повышенной утомляемостью и потемнением кожных покровов на фоне симптомов гиперандрогении.
Для женщин с гиперандрогенией при классической форме дефицита 21-гидроксилазы характерна вирилизация наружных половых органов, аменорея, выраженная алопеция и гирсутизм. При несвоевременной диагностике пациентки с женским кариотипом могут иметь мужской фенотип. У мужчин данный синдром проявляется бесплодием и акне средней и тяжелой степени тяжести.
Также выделяют неклассическую форму АГС, второе ее название — постпубертатная. Она имеет наиболее благоприятный прогноз, при этом специфические клинические симптомы отсутствуют. Как правило, синдром дебютирует в пубертатном возрасте или после перенесенной стрессовой нагрузки, такой как беременность, выкидыш на раннем сроке, медицинский аборт и др. У женщин эта форма проявляется в виде угревой сыпи, позднего наступления менархе (15–16 лет). При этом цикл неустойчив, интервалы могут достигать до 45 дней и более. Менструальные выделения скудные, молочные железы слабо развиты. Чем позже дебютирует заболевание, тем меньше выражены фенотипические проявления этого синдрома.
Основным осложнением АГС является бесплодие, и, чем раньше произошла манифестация заболевания, тем меньше вероятность беременности в будущем. При неклассической форме заболевания на ранних сроках беременности возникают самопроизвольные выкидыши, а во время родов возможно развитие истмико-цервикальной недостаточности.
Диагностика осуществляется лабораторными и инструментальными методами.
Уровень 17-ОН-прогестерона. Высокая концентрация 17-гидроксипрогестерона, являющегося предшественником кортизола — ключевой признак недостаточности 21-гидроксилазы.
Содержание андростендиона в сыворотке крови. Повышенные показатели этого высокоспецифичного маркера подтверждают усиленную секрецию предшественников мужских половых гормонов.
Уровень АКТГ в крови. Для классических форм заболевания характерна компенсаторная гиперсекреция адренокортикотропного гормона передней долей гипофиза.
УЗИ яичников. В корковом веществе определяются фолликулы на разных стадиях созревания, не достигающие преовуляторных размеров.
Молекулярно-генетическая диагностика. Исследование гена CYP21 и определение его мутаций, что позволяет определить форму заболевания и прогноз течения синдрома, а также вероятность рождения больного ребенка.
Для сольтеряющего варианта АГС характерна повышенная концентрация ренина в плазме крови.
Читайте также: