
Аутистические расстройства синдром ретта доклад
Обновлено: 05.07.2024
Синдром Ретта - этиология, клиника, диагностика
Синдром Ретта является необычным нарушением развития мозга, с напоминающими аутизм или нейродегенеративное заболевание признаками, но в действительности не является ни одним из них. Синдром Ретта представляет собой характерный комплекс клинических проявлений, включающих раннюю психомоторную регрессию с аутистическими проявлениями, замещение целенаправленных действий рук стереотипными движениями, атаксией и апраксией при ходьбе и приобретенной микроцефалией (Hagberg, 1989). В таблице 4.4 представлены международные критерии диагностики (Rett Syndrome Diagnostic Criteria Work Group, 1988; Trevathan и Naidu, 1988). За редким исключением данное заболевание встречается только у девочек (Zoghbi, 1988, Hagberg, 1989).
а) Распространенность. Распространенность синдрома Ретта в Швеции и западной Шотландии, составляет 1 на 10000 и 1 на 18000 девочек (Kerr и Stephenson, 1985; Hagberg, 1993; Bienvenu et al., 2006).
б) Патогенез. Синдром Ретта приблизительно в 80% случаев связан с мутациями гена МЕСР2, и длительное время считалось, что мутация данного гена является причиной синдрома Ретта. Тем не менее, фенотипические проявления мутаций гена МЕСР2 разнообразны и включают задержку умственного развития с припадками или без них, фенотип, подобный синдрому Ангельмана, и аутизм (Zoghbi, 2005). Как минимум еще один ген (CDKL5) связан с развитием судорожного варианта заболевания. Ген МЕСР2 является ингибитором фактора транскрипции, способным отключать несколько важных для развития головного мозга генов, а в связи с экспрессированием в различных типах клеток и органов— влиять на соматическое развитие в целом. В этой связи, представляется вероятным, что мутация гена МЕСР2 при синдроме Ретта является частью последовательной цепи событий, приводящих к развитию ряда сцепленных с Х-хромосомой нарушений развития нервной системы.
В настоящее время синдром Ретта представляется скорее патологией развития, а не дегенеративным заболеванием (Naidu, 1997). Структурные аномалии мозга выражены слабо и включают маленький размер мозга с плотно расположенными нейронами и снижением клеточных процессов. В подавляющем большинстве случаев заболевание не имеет наследственного характера, несмотря на то, что выборочное поражение девочек предполагает генетическое происхождение синдрома.

в) Клинические проявления. Течение синдрома Ретта имеет необычный характер. Заболевание начинается как прогрессирующее состояние с более или менее стремительной деградацией с утратой ранее полученных навыков.
Течение заболевания может быть разделено на четыре стадии. Дебют клинических симптомов приходится на возраст от шести месяцев до трех лет, в большинстве случаев заболевание манифестирует до 18 месяцев. Изначально развитие ребенка может не отличаться от нормы, но у больных девочек часто с рождения отмечается гипотония и слегка замедленное развитие (Einspieler et al., 2005).
Небольшое количество пациентов страдает от приступов ярости, тревоги, смущения и беспорядочной гиперактивности. При наличии аутистической или подобной аутистической фазы данное состояние может длиться от одного месяца до нескольких лет. Обычно к достижению школьного возраста (или не позднее пубертатного периода) аутистические симптомы начинают убывать, но не во всех случаях. По имеющимся данным, у большинства аутистов, вне зависимости от причины заболевания, присутствует одинаковый характерный тип развития.
Бруксизм и гипервентиляция являются типичными проявлениями синдрома Ретта и иногда интерпретируются как признаки чрезвычайной тревожности, что не подтверждается опытом.
На МРТ выявляется уменьшение объема мозга, преимущественно за счет белого вещества, уменьшение объема хвостатого ядра и среднего мозга и нормальное строение извилин (Reiss et al., 1993).

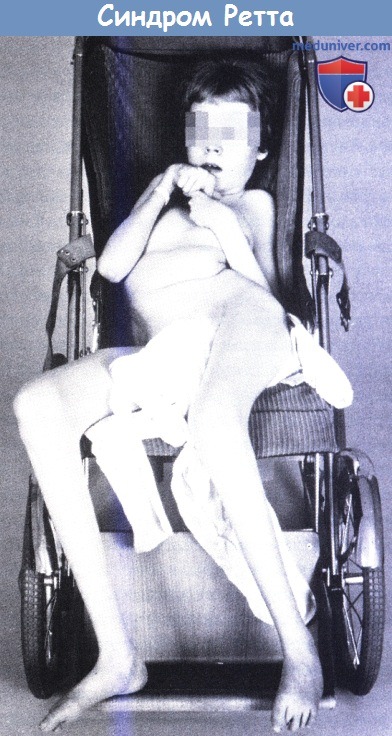
Синдром Ретта у 15-летней девочки.
Характерные стереотипные движения кистей, выраженный сколиоз и атрофия нижних конечностей.
д) Диагностика. Диагноз синдрома Ретта до сих пор устанавливается на основании анамнеза и клинических проявлений, а обнаружение мутации гена МЕСР2 во многих случаях служит подтверждением (Huppke и Gartner, 2005).
Диагностические критерии синдрома Ретта приведены в таблице ниже. У многих девочек с синдромом Ретта на ранних стадиях заболевания отмечаются аутистические проявления без четко выраженных неврологических отклонений. Поэтому в возрасте до трех лет часто ставится диагноз аутизма. Диагноз синдрома Ретта учитывается во всех случаях выявления симптомов аутизма у девочек в очень раннем детском возрасте. В исследовании Witt-Engerstrom и Gillberg (1987) девочек с синдромом Ретта в 80% случаев изначально предполагался аутизм или проявления аутизма, но на основе имеющихся данных о распространенности было установлено, что в 1/3-1/2 случаев выявления симптомов аутизма в течение первых лет жизни в итоге определялся симптом Ретта.
е) Дифференциальная диагностика синдрома Ретта. В основном необходима дифференциальная диагностика с синдромом аутизма. Нейронный восковидный липофусциноз новорожденных с движениями рук, имитирующими вязание, описанный Santavuori, может иметь сходство с синдромом Ретта, но редко встречается за пределами Финляндии (Santavuori et al., 1973; Santavuori 1988). Дефицит орнитинтранскарбомилазы также ошибочно может приниматься за синдром Ретта. Синдром Ангельмана может иметь сходство с синдромом Ретта в связи с судорожной атаксией, наблюдаемой в обоих случаях помимо проявлений аутизма, задержки умственного развития и припадков. Исследование хромосом в сложных случаях позволяет дифференцировать заболевания.

ж) Лечение. Лечение синдрома Ретта неэффективно. При попытке применения бромокриптина и налоксона положительных результатов получено не было. Пациентам необходима физиотерапия и внимательное отношение к деталям обыденной жизни.
Стимуляторы показаны девочкам с хорошей реакцией на лечение в течение раннего периода отмены препарата. Важной частью терапии синдрома Ретта является ортопедическое лечение для предупреждения развития или уменьшения выраженности сколиоза.
Лечение поведенческих/психиатрических отклонений, вызванных синдромом Ретта, требует знания естественного течения заболевания, чтобы такие симптомы как аутизм и ночной смех не интерпретировались ошибочно как специфические психологические отклонения или проблемы общения. Восприятие речи при синдроме Ретта чрезвычайно снижено. Общение осуществляется с помощью зрительного контакта и жестов. Некоторые функции рук могут быть восстановлены при длительной ежедневной тренировке каждой руки в отдельности.
При применении бромокриптина (20 мг/кг в сутки) были достигнуты некоторые положительные результаты (Zappella et al., 1990), но для подтверждения эффективности необходимо проведение двойных слепых пла-цебо-контролируемых исследований. Результаты применения налоксона неоднозначны.
з) Исход. Отдаленные исходы синдрома Ретта известны только частично. Продолжительность жизни относительно увеличена, некоторые пациенты достигают возраста 80 лет и более. Подавляющее большинство (практически все) пациенты имеют чрезвычайно выраженные неврологические и/или умственные отклонения и зависят от других людей практически во всех областях повседневной жизни. В большинстве случаев клиническая картина осложняется эпилепсией, запорами, сколиозом, прогрессирующими двигательными (и вазомоторными) отклонениями. Психиатрические/по-веденческие отклонения могут являться предметом озабоченности в детском и иногда в подростковом возрасте, но обычно они в меньшей степени затрагивают пациентов старшей возрастной группы.
Ожидайте
Специалист свяжется с Вами сразу в рабочее время с
Пн - Пт с 10:00 - 19:00 МСК
Перезвоните мне

Ваш персональный менеджер: Екатерина
Ответственная и отзывчивая! 😊
Ожидайте
Специалист свяжется с Вами сразу в рабочее время, ежедневно с 10:00 - 19:00 МСК
Перезвоните мне

Синдром Ретта – это генетическое заболевание, сопровождающееся тяжелыми психоневрологическими симптомами.
Бесплатные занятия с логопедом
Бесплатный курс ИКТ для детей
Синдром Ретта – это генетическое заболевание, сопровождающееся тяжелыми психоневрологическими симптомами. Диагностика его затруднительна: оно практически никогда не обнаруживается внутриутробно, а после рождения проявляется не ранее, чем через 6 месяцев. Своему носителю оно грозит глубоким слабоумием, двигательными ограничениями, дезадаптацией в социуме.
ИСТОКИ ЗАБОЛЕВАНИЯ
В масштабном формате о расстройстве заговорили в 1983 году благодаря шведскому ученому Бенгту Хагбергу. В это время он со своей группой изучал 35 подобных между собой случаев в 3 разных странах: в Португалии, Франции и Швеции.
Однако Хагберт не является первооткрывателем синдрома. Впервые его обнаружил педиатр Андреас Ретт, имя которого носит заболевание. Он наблюдал за двумя девочками, имеющими одинаковые симптомы. Их он заметил в очереди на прием. Они сидели на коленях у матерей, а те держали их за руки. Девочки раскачивались как маятники, а затем внезапно обе начали совершать стереотипные движения руками. Дети застыли в одном положении, отстраненные от окружающего мира. Взгляд был направлен в одну точку. Поражала их синхронность в движениях и поведении.
В своих письменных архивах врач отыскал подобные истории болезни, а затем отправился в Европу, чтобы разыскать и там таких же пациентов. В 1966 он сделал первые публикации своих исследований, которые, однако, не вызвали особого интереса.
Зафиксированную им болезнь Ретт назвал синдромом атрофии мозга. Сначала ее считали проявлением аутизма или шизофрении, и только лишь в 1983 году вывели в отдельную нозологическую единицу.
В настоящее время синдром относят к категории довольно редких генетических заболеваний. Он встречается с частотой случаев 1 на 15000. Причиной его называют мутацию гена МЕСР2. Этот ген отвечает за синтез определенного белка, влияющего на развитие мозга. В норме этот белок, спустя некоторое время после рождения, должен подавляться другими генами, чтобы обеспечить нормальное развитие мозга.
Если же ген МЕСР2 мутирован, то белок инактивируется не полностью, что вызывает аномальное мозговое созревание, и провоцирует развитие синдрома Ретта.
Обычно мутирующий ген располагается в Х хромосоме, потому заболеванием страдают преимущественно девочки.
ПОЧЕМУ МАЛЬЧИКИ НЕ БОЛЕЮТ
У мальчика Х-хромосома одна. Если она имеет мутационный ген, значит, выпадает из работы полностью, и ее нечем заменить. Такие малыши мужского пола, как правило, погибают еще внутриутробно, так и не родившись. Поэтому синдром Ретта у мальчиков встречается крайне редко.
Но, несмотря на такую особенность заболевания, очень редко, но все-таки мальчики с подобным синдромом выживают. Это может быть связано с тем, что не все гены в Х-хромосоме подвергаются мутации. Из-за этого заболевание развивается не столь остро.
Другая причина – наличие у мальчика синдрома Клайнфельтера. При этом наблюдается полисомия половых хромосом, то есть их набор составляет ХХУ. И, если одна Х-хромосома имеет патологический ген, то вторая может регулировать синтез белка и дарить мальчику возможность жизни. Получается такая же картина, как и у девочки.
КАК РАЗВИВАЕТСЯ ЗАБОЛЕВАНИЕ
Синдром Ретта у детей – довольно коварное заболевание. При рождении оно практически не проявляет себя. Первые его симптомы появляются в период от 6 мес. до полутора лет. Однако некоторые, еле заметные признаки, в первом полугодии все-таки имеются. Но они настолько ничтожны, что не привлекают внимания.
В 1 год и 7 мес. она перестала узнавать родителей и, казалось, не нуждалась в них. Весь день проводила за одним однообразным занятием: кидала мяч или катала коляску. Часами ходила по кругу, пока ее не останавливали или она запиналась. Такое стереотипное поведение носит название полевого, когда действие затягивает больного, и он не может ничего сделать.
В четыре года к симптомам присоединились эпилептоидные припадки. Однако по достижении школьного возраста девочка находилась на домашнем обучении, и делала некоторые успехи.
12–6 лет – это был период ремиссии, когда болезнь практически не беспокоила. Но с 16 лет появились новые, более глубокие проблемы, связанные с костными деформациями и болезнями внутренних органов. Одна нога девочки была короче другой почти на 10 см, что не могло не препятствовать ходьбе. В 20 лет она весила всего 24 кг с ростом 158 см.
Обычно СР протекает в 4 стадии.
Первая стадия, которая, как правило, стартует с 6 месяцев до полутора лет, проявляется нарастанием раздражительности и лабильностью настроения у ребенка. Эпизоды плача и психомоторного возбуждения сменяются все большей пассивностью. Малыш бесцельно передвигается по комнате, пропадает интерес к игрушкам. Но контакт с матерью сохраняется.
Вот как описывает женщина поведение своей дочери на заре заболевания: она кричала целый день без остановки, билась головой о стены, не могла уснуть. Что бы мы ни делали, она не успокаивалась. Это был настоящий ад. Но больше угнетало то, что ни один врач не мог поставить вразумительный диагноз.
Развивается диспропорция головы и конечностей по отношению к телу. Они становятся несоизмеримо маленькими. Замедляется рост, и снижается тонус мышц.
Вторая стадия, длящаяся несколько лет, отличается пестротой симптомов. Сразу обращает на себя внимание снижение интеллектуальных способностей, развивается умственное слабоумие. Происходит регресс практически всех полученных навыков. Речь полностью исчезает или переходит в степень эхолалии – механического повторения услышанного.
Приобретенные двигательные навыки, предметно-ролевое поведение теряются и замещаются двигательными стереотипами. Характерный симптом: многочисленно повторяющиеся движения, напоминающие мытье рук. Кроме этого, ребенок постоянно заламывает или потирает их, размахивает ими, хлопает в ладоши. Сжатие пальцев рук вполне нормально в 4 месяца, но в более позднем возрасте говорит об остановке развития. Малыш утрачивает хватательный рефлекс, не способен производить вращательные движения руками.
Постепенно двигательная активность сходит на нет. Нарушается походка, ребенок ходит, не сгибая коленей.
Третья стадия длится 10 лет и более, характеризуется она развитием стойкого, глубокого слабоумия, вплоть до идиотии. Наблюдается полная потеря способности говорить и понимать обращенную к ребенку речь. Появляется тремор всего тела, отягчающий движения. Усиливаются судорожные припадки.
Четвертая, конечная стадия – это период усугубления ранее проявляемых симптомов. Стойкая утрата умственных способностей, двигательных навыков, развитие мышечных дистрофий, приводящих к полному обездвиживанию.
Продолжительность жизни таких больных в среднем колеблется до 30 лет, хотя известны случаи, когда они доживали и до 50-летнего возраста.
САМЫЕ ЧАСТЫЕ СИМПТОМЫ РАССТРОЙСТВА
Типичные симптомы для синдрома Ретта – мышечные и двигательные нарушения. Мышцы находятся в гипертонусе или же, наоборот, теряют его. В этом случае у ребенка развивается неправильное положение тела, прогрессирует частичные параличи и нарушение координации. Например, девочки скрещивают ноги во время ходьбы.
Синкинезии – патологические сокращения мышц, возникают вслед за произвольным движением: простая улыбка способна вызвать резкий взмах ногой. Такое явление постепенно приводит к повреждению суставов, сухожилий и связок, провоцирует ортопедические нарушения. Последние проявляются во всевозможных деформациях и также очень часто сопровождают таких детей. Среди них выделяют вывих тазобедренного сустава, провоцируемый малой подвижностью.
Сколиоз – боковое искривление позвоночника, который провоцирует массу проблем у таких пациентов: деформации суставов и костей, боли во время ходьбы, в стоячем или сидячем положении, утрата способности передвигаться. Сколиоз грудного отдела вызывает легочную недостаточность. Появляются также проблемы с пищеварением.
У детей с синдромом Ретта наблюдается повышенное слюнотечение. Но это происходит не из-за избытка количества слюны, а потери способности сглатывать ее.
Нарушение питания может развиваться из-за частых приступов тошноты. Она появляется на любые аспекты питания: на определенный продукт, его температуру, на способ приготовления. Так, ребенок способен отрицательно реагировать на пищу, поданную кусочками, или на комочки в блюде.
Постоянная тошнота провоцирует отказ от питания, а значит, потерю в весе.
Плохое сглатывание слюны, которая регулирует кислотность в желудке, и повышенное внутрибрюшное давление вызывают желудочно-пищеводный рефлюкс, то есть забрасывание содержимого желудка в пищевод. Это чревато такими последствиями, как воспаление стенки пищевода, респираторные инфекции.
Малоподвижный образ жизни, неврологические расстройства, неправильное питание провоцируют возникновение запоров у детей с синдромом Ретта. Они носят тяжелый характер, поскольку способны вызывать закупорку кишечника и сильные боли.
Повышенное слюнотечение, тошнота, рефлюкс снижают потребление ребенком пищи и даже развивают на нее негативную реакцию. В результате этого ребенок теряет в весе. Этот процесс стоит строго контролировать, поскольку он чреват истощением.
Другое тяжелое расстройство связано с работой дыхательной системы, развивающееся вплоть до приступов апноэ. Это явление настолько часто среди детей с синдромом, что нередко стает причиной их гибели.
Важными патогномоничными признаками синдрома считаются проявления аутизма. Именно из-за них заболевание изначально считали одной из форм этого расстройства, а в настоящее время относят к болезням аутистического спектра.
Аутистические признаки проявляются в отстранении от окружающего мира, в том числе и от родственников. Ребенок замыкается в себе, может не откликаться, когда его зовут. Предпочитает одиночество. Дети боятся чужих людей и непривычных ситуаций.
Лицо такого ребенка становится похожим на каменное. Взгляд блуждающий или устремлен в одну точку. Поведение часто непредсказуемо: случаются приступы неутомимого смеха или плача. Склонны к самоповреждениям: царапают кожу, кусают пальцы, вырывают волосы.
НЕТИПИЧНАЯ КАРТИНА
Наряду с типичной формой заболевания, описанной выше, встречаются и атипичные формы. Они имеют свои особенности, от которых зависит тяжесть заболевания.
- Zapella – форма синдрома с неярко выраженными признаками. Речь частично сохранена, умеренно выражен сколиоз, умственная отсталость средней степени тяжести. Физически развиваются нормально.
- Hanefeld – в клинической картине преобладает раннее развитие судорожных приступов. Часто они случаются даже до появления умственной деградации.
- Rolando – на первый план выходят признаки задержки психомоторного развития. Ребенок теряет возможность передвигаться, нарастает стереотипия движений, его беспокоят дыхательные нарушения.
Синдром Ретта – сложное генетическое заболевание. Прежде всего, его сопровождает полная умственная деградация и психоневрологические нарушения, влекущие за собой многочисленные патологии других систем организма.
К сожалению, в мире еще не существует способа кардинального искоренения болезни, хотя ученые ведут постоянные разработки в этом направлении.
Лечение синдрома сводится к трем основным направлениям. Медикаментозная терапия назначается для купирования судорожных припадков и стимуляции работы головного мозга.
Диетотерапия включает в себя контроль массы тела, употребление в пищу высококалорийных, витаминизированных продуктов.
Однако наибольшее внимание уделяется реабилитационным мероприятиям, направленным на укрепление опорно-двигательного аппарата и поддержание умственного, психомоторного развития.
Важно сохранить комплексный, всесторонний подход к проблеме. Такие дети нуждаются в постоянной поддержке со стороны взрослых и веры в них. Сотрудничество с ними, как с полноценной ячейкой общества, способствует их лучшей адаптации в социуме и более благоприятному развитию.

Синдром Ретта — крайне редкое наследственное заболевание, которое возникает у девочек преимущественно в раннем возрасте, характеризующееся регрессом психомоторного развития, поведением, схожим с аутистическим, потерей навыков целенаправленного движения руками и эпилептическими припадками [1]. Частота встречаемости, как правило, составляет 1:15000 [3].
Впервые данное заболевание описывается в литературе второй половины ХХ века, когда австрийский педиатр Адресасс Ретт при исследовании группы девочек из разных городов Европы дал описание синдрому, который проявляется прогрессирующей потерей психических и двигательных навыков [1].
Синдром Ретта возникает в результате нарушения развития головного мозга, причиной чего является мутация в гене транскрипции МЕСР2, связанного с Х хромосомой, находящейся в локусе Хq28 [2]. На сегодняшний день выявлено 8 мутаций данного гена. Предполагается, что ген МЕСР2 контролирует процессы развития ЦНС. Важно отметить, что мутации данного гена могут встречаться и у мальчиков, однако такие особи в популяции нежизнеспособны и умирают в первые часы жизни [1,2].
Согласно последним результатам нейрофизиологических исследований, при синдроме Ретта имеются нарушения со стороны синаптической организации, а также нарушения дофаминового обмена, обнаруживаются атрофические изменения в ростральных отделах коры головного мозга (КГМ) , которые возникают на ранних этапах онтогенеза [2]. Атрофические изменения также обнаруживаются в nucleus caudatus, substantia nigra. Нарушения в развитии КГМ проявляются грубыми нарушениями функций лобных долей мозга с выпадением их организующих влияний. Происходит деафферентация и растормаживание иерархически нижестоящих отделов коры и подкорковых структур, что ведет к регрессу психомоторного развития с возрастом [1,2].
Интервал проявления заболевания — от 6 до 20 лет, в крайне редких случаях первые симптомы выявляется в возрасте до 3 месяцев. Важным патогномоничным признаком является нормальное психомоторное развитие до начала заболевания [2]. Неспецифическими симптомами являются: врожденная гипотония, гипомимия, отставание в приобретении моторных навыков, слабое сосание, монотонный безутешный плач, эмоциональная ригидность, проблемы при фиксации взгляда [3].
Клинически синдром Ретта подразделяют на четыре стадии [2].
1. Первая стадия или стадия стагнации. Продолжительность от 2 до 4 месяцев, редко больше. В этой стадии происходит остановка психомоторного развития, замедление темпов роста головы, костей и стоп. Появляется мышечная гипотония, пропадает интерес к играм и окружающему миру, нарушается зрительное сосредоточение. На данной стадии клинический диагноз поставить невозможно [1].
3. Третья стадия (псевдостационарная). Характеризуется относительной стагнацией развития заболевания. Происходит уменьшение симптомов аутистического поведения, проходят приступы беспокойства, нормализуется сон. В данной стадии клинически превалируют нарушения гнозиса и праксиса, сохраняются стереотипии в руках и отсутствие целенаправленной мануальной деятельности. Выявляются признаки нарушения проведения нервного импульса по пирамидному пути, статическая и динамическая атаксия, мышечная дистония, тремор [1,3].
Для данного этапа развития заболевания характерны 3 разновидности пароксизмальных состояний: эпизоды гипервентиляции, синкопальные состояния и эпилептические приступы [3].
4. Четвертая стадия или стадия поздних двигательных нарушений. Данная стадия характеризуется прогрессированием развития моторных расстройств и, как правило, проявляется ближе к 10 годам. Усугубляются парезы, нарастает выраженность спастико-амиотрофического синдрома. Хорошо заметна мышечная ригидность в нижних конечностях. Увеличивается степень сколиоза, кифосколиоза. К 15 годам ребенок не способен самостоятельно ходить [1].
Таблица 1 | Диагностические критерии типичного синдрома Ретта [1]
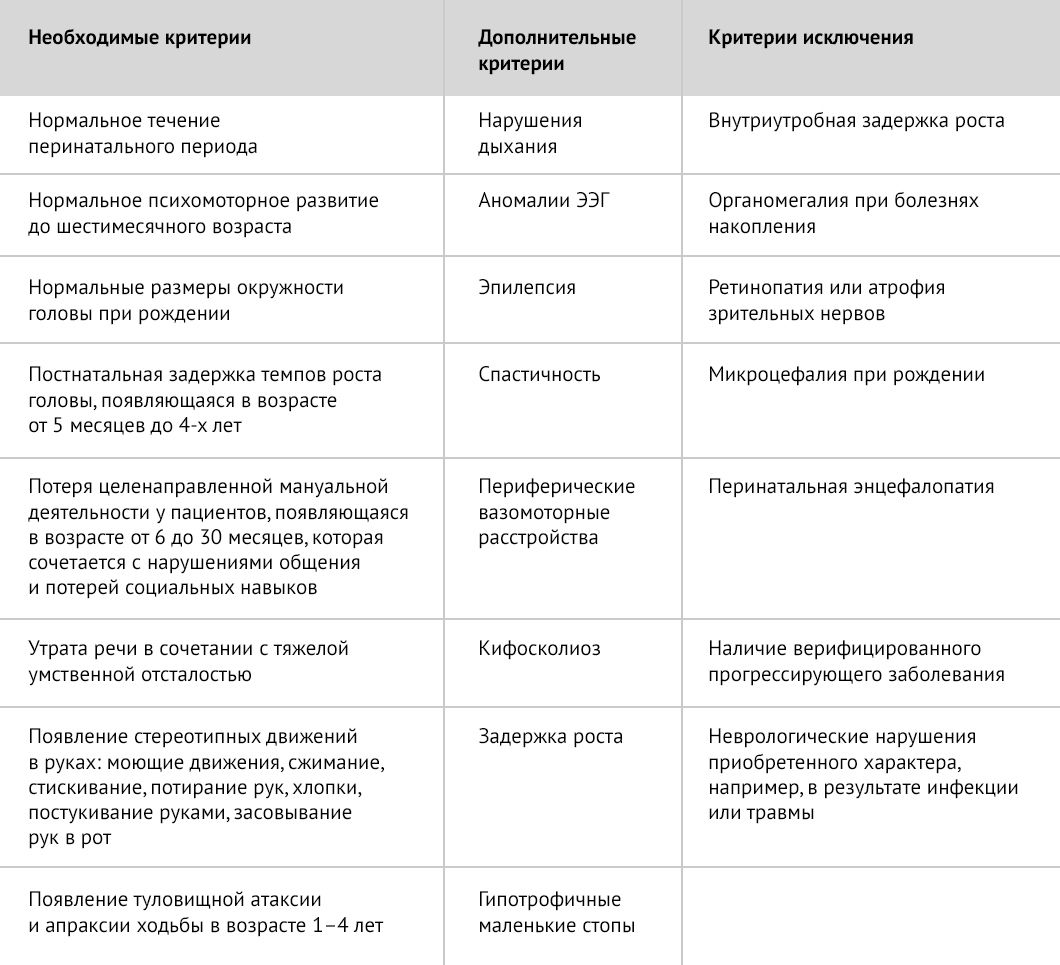
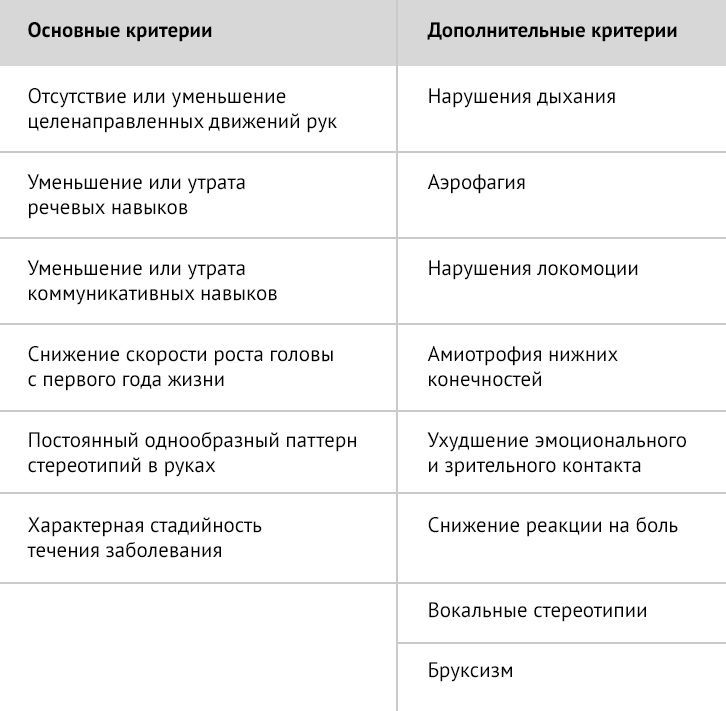
Таблица 2 | Диагностические критерии атипичного синдрома Ретта [1]
Главной задачей в лечении синдрома Ретта является контроль эпилептических припадков. Лучшие препараты для этих целей — карбамазепин, ламотриджин, топирамат [1].
.
В отечественной детской неврологии первый расширенный обзор по синдрому Ретта приведен И. А. Скворцовым и соавт. (1992); в детской психиатрии первые собственные клинические наблюдения синдрома Ретта даны В. М. Байтной, Н. В. Симашковой и соавт. (1992, 1993, 1994). Синдром Ретта впервые введен в круг первазивных нарушений развития в МКБ-10 (1994).
Происхождение синдрома Ретта остается недостаточно выясненным. К настоящему времени большинство исследователей предполагают, что синдром Ретта — не нейродегенеративное прогрессирующее мозговое поражение, а скорее генетическое нарушение развития мозга, и связывают синдром Ретта с нарушениями в Х-хромосоме. До 1990 г. считалось, что синдром Ретта поражает только девочек [Annvert M. et al., 1990]. В последние годы появились единичные публикации [Philippart М., 1990; Zappella M., 1990], в которых представлено описание лиц мужского пола с синдромом Ретта.
Распределение по полу. Существует представление, что синдром Ретта возникает у девочек, однако в литературе описаны отдельные случаи расстройств, сходных с синдромом Ретта, у мальчиков, и пока вряд ли можно говорить, что синдрома Ретта не бывает у мальчиков [Hagberg В., Gillberg Ch., 1993].
В 4 /5 случаев у матерей детей с синдромом Ретта отмечены нормально протекавшие беременность и роды, в 1 /5 случаев наблюдалась легкая пре- и перинатальная патология. Раннее (до 6—16 мес жизни, редко позже) развитие умственной и моторной деятельности в половине случаев было нормальным, в другой половине выявлена легкая (на 2—3 мес) задержка в становлении крупных моторных актов. Отмечаются экстравертированность, эмоциональная живость, социальная направленность у этих детей.
В I стадии (аутистической) наблюдается замедление психического развития, снижение интереса к игровой деятельности и окружению, мышечная дистония, замедление роста головы. Длительность стадии от нескольких месяцев до 10 мес и более. На этом этапе синдром Ретта трудно отличим от аутистических расстройств.
повторам через разные промежутки времени. В ряде случаев при подобном нарушении дыхательного ритма обнаруживается заглатывание воздуха с возможным пневматозом кишечника. Апогей этой стадии болезни у разных детей формируется в периоде от нескольких месяцев до нескольких лет (1—2 года), редко позже.
III стадия (псевдостационарная) характеризуется наличием явной деменции. На этом этапе насильственные движения в кистях рук протекают с меньшей силой, частотой и напряжением, приобретают прерывистый характер. Тогда уже у ряда детей появляется крупноразмашистый тремор рук, головы, усиливающийся при выполнении направленных движений, вернее, попыток к их выполнению. У ряда больных сохраняются расстройства дыхания. В половине случаев от числа наблюдаемых больных возникают эпилептиформные приступы (от абсансов, малых приступов до развернутых больших припадков), а также в редких случаях бывают приступы по типу вздрагиваний, которые в некоторых случаях сопровождаются ознобоподобными симптомами, дрожью всего тела, наступающими во сне, независимо от времени сна (дневного и ночного). В ряде случаев гиперкинез в группах мышц плечевого пояса протекает по типу учащенных разрядов, что отдаленно напоминает приступы фокальной эпилепсии, без потери сознания. Отмечается практически полная утрата речи. Лишь у отдельных детей сохраняются единичные слова, слоги.
Психопатологические исследования выявляют на этом этапе тотальный психический распад, глубокие расстройства личности, невозможность к обучению. У отдельных детей аутохтонно в этом периоде относительной стабилизации смягчаются обнаруживаемые моторные расстройства (хотя насильственные движения в кистях рук в стертой форме сохраняются). Возникает способность к усвоению некоторых элементарных навыков, оживляется интерес к игрушкам, в отдельных случаях к книгам, картинкам в них. Сохраняется истощаемость. В эти периоды можно наблюдать смены настроения от слегка приподнятого к раздражительно-дисфорическому.
Обнаружен синдром Ретта с неполным проявлением отдельных стадий болезни: с острым началом регресса, более короткими по времени другими стадиями болезни и быстрым распадом моторных функций, утратой ходьбы.
Есть, напротив, течение синдрома Ретта со спутанными стадиями, молниеносным присоединением эпилептических приступов и вместе с тем как бы приостановкой в последующем злокачественного течения.
Описаны случаи синдрома Ретта, коморбидные с нарушениями по 21-й, 13-й и другим хромосомам; делецией 13-й хромосомы, q 12.1 и q 21.2, обнаруженных с помощью цитогенетического анализа [Herder G. A., 1996].
Все эти случаи синдрома Ретта отнесены к так называемым атипичным его формам, существование которых к настоящему времени является признанным фактом. Знание этих вариантов синдрома Ретта очень важно для диагностики, прогноза и подбора терапии. В диагностическом аспекте всегда трудно разграничивать синдром Ретта с детским аутизмом, в особенности при затяжной аутистической стадии. Синдром Ретта с быстрым ранним появлением эпиприступов трудно различим с эписиндромом и эпилепсией. Возникает необходимость тщательной дифференциальной диагностики с мукополисахаридозами, при которых нередки похожие симптомы типа стереотипии в моторике и умственного недоразвития. На поздних стадиях синдром Ретта нуждается в дифференциации с УМО разного генеза, дефектными, постприступными состояниями при инфантильном психозе (ранней детской злокачественной шизофрении). На последней остановимся более подробно.
Общими для больных с синдромом Ретта и инфантильным психозом были следующие феномены: аутохтонное начало заболевания; возраст начала болезни в 8—30 мес жизни; быстрое присоединение регресса всех форм психической деятельности; некоторое ослабление болезни в середине второго физиологического возрастного криза.
Различными в этих двух группах патологических состояний были следующие феномены: половой состав групп; глубина проявлений регресса (тяжелый регресс моторной, речевой сфер деятельности при синдроме Ретта; менее выраженный регресс этих же сфер при инфантильном психозе).
Больным с синдромом Ретта свойствен более тотальный распад речи. Для больных с инфантильным психозом оказались более характерными нарушения в ассоциативном процессе, тенденция к эгоцентрической речи и частичное сохранение ее понимания. При синдроме Ретта в первую очередь страдала моторная сторона речи, сенсорная сохранялась более длительный срок. Однако в дальнейшем наблюдался полный распад речи, чего никогда не обнаруживалось при инфантильном психозе.
Больным с инфантильным психозом присуще переслаивание моторных формул, соответствующих возрасту, с более ранними архаическими моторными стереотипиями, характерными для первых недель жизни ребенка, при сохранении способности к эволютивному усложнению моторных актов. У детей с синдромом Ретта наряду с появлением физиологически более древних моторных формул наблюдались нарушения моторики, свойственные конечным этапам тяжелых органических заболеваний ЦНС (грубый прогрессирующий распад моторных формул с мышечными атрофиями, нарушениями дыхания, жевания, глотания, удерживания предметов, утратой ходьбы).
Эмоциональная сфера у больных с инфантильным психозом поражалась более грубо, распадались связи с родными, отставало формирование эмоциональных контактов. У детей с синдромом Ретта на фоне тотального распада всех сфер деятельности длительнее сохранялись эмоциональная адекватность, привязанности.
В структуре аутизма при инфантильном психозе наиболее специфичным оказался диссоциированный характер функционирования всех сфер деятельности ребенка. При этом нарушалась иерархия всех функционирующих систем, без физиологического, как в норме, вытеснения ранних форм реагирования более поздними, высшими — в когнитивной, речевой, моторной и эмоциональной сферах. При синдроме Ретта в основе аутизма, по-видимому, лежат как неуточненные причины, так и насильственная деятельность, которая ведет к затормаживанию всех других видов направленной деятельности. В этих случаях возникающее отрешение с аутизацией в поведении лишь поверхностно напоминает аутизм при инфантильном психозе и с течением болезни аутистические проявления сглаживаются. Таким образом, сравнительное клинико-психопатологическое изучение больных с наиболее тяжелыми формами аутизма показало, что всем аутистическим синдромам в круге инфантильного психоза и синдрома Ретта оказались свойственны: аутистическое поведение, качественное повреждение вербальной и невербальной коммуникации, моторные расстройства. Структурное сходство перечисленных психопатологических проявлений позволяло во всех случаях диагностировать первазивные расстройства (по МКБ-10, 1994). Приводим следующее наблюдение.
Больная Д., 1988 года рождения. Родилась от нормально протекавшей беременности; роды в срок. Психомоторное развитие на первом году жизни близко к возрастной норме. Руление с 4 мес, лепет с 8 мес, первые слова — после года. Приблизительно после полутора лет пропало общение с матерью, стала безразличной, исчезла речь. В кистях рук появились потирающие движения, стала бить себя по подбородку, во время ходьбы пошатывалась. Остановилась в психическом развитии, перестала играть, игрушки в руках не удерживала. Постепенно перестала ходить, предпочитала сидеть. После 3 лет появились эпилептические припадки.
В неврологическом статусе: череп микроцефальной структуры, асимметрия оскала. Кисти ластовидные. Варусная установка стоп. Проксимальная гипотония мышц верхних и нижних конечностей. Слюнотечение, нарушение жевания. Подволакивает обе ноги. Не захватывает и не удерживает предметы в руках, нарушена тонкая моторика кистей рук. Общая дискоординация движений. Сенсомоторная дисфазия. На ЭЭГ — очаги эпиактивности.
Проведен курс терапии церебролизином (метамерное обкалывание по Осипенко — Скворцову).
Данные психологического обследования: интеллектуальное развитие неравномерное, соответствует возрасту 7—12 мес. Глазное слежение сохранено. Слуховое внимание недостаточное, истощаемое. Экспрессивная речь на лепетном уровне. Понимание отдельных бытовых просьб на уровне 12 мес. Игрушки захватывает, но не удерживает. Картинками не интересуется, не узнает на них отдельные предметы.
Катамнез: 7 лет 4 мес. Психический статус (после третьего курса терапии церебролизином). Внимание можно привлечь. Обращенную речь элементарно-бытового уровня понимает. Возникают попытки к элементарному общению на интонационно-фонематическом уровне, произносит несколько первых слогов, слов. Менее истощаема, улучшилось слежение за предлагаемыми картинами. Может следить за детскими передачами по телевидению.
Психологическое обследование. Обнаруживается неразвитость психических функций: крупная моторика на уровне 2 лет, мелкая — на уровне 12 мес. Речи нет. Социальное общение — на уровне 18 мес, интеллектуально-мыслительные операции — 8—12 мес, игра — 12 мес. Освоены туалетные навыки. Формируется индивидуальное отношение к бабушке, оживилась эмоциональная сфера.
Логопедическое обследование: остановка речевого развития.
Генетическая консультация. Диагноз синдрома Ретта подтвержден (Х-сцепленное рецессивное наследование). На фоне проводимого лечения отмечается положительная динамика.
Неврологическое обследование. Мышечный тонус диффузно снижен. Сухожильные рефлексы D>S. Стереотипные движения в кистях рук в виде постукивания и похлопывания ладонью о ладонь. Ходит самостоятельно.
ЭЭГ-исследование. Изменения резидуально-органического характера; эпилептических знаков нет.
Консультация логопеда: сенсомоторная дисфазия.
Заключение. Состояние больной можно отнести к стационарной стадии синдрома Ретта. Его определяют тяжелое умственное недоразвитие, недоразвитие речи, остаточные движения в кистях рук. Сниженная активность и ограниченность в общении объясняются не столько аутистическими формами поведения, сколько тяжелым умственным недоразвитием. Сохраняются гипотония, кифосколиоз, бедность побуждений.
Особенность данного наблюдения состоит в формировании частичного восстановления утраченных функций (ходьбы, удержания предметов, глотания, жевания, навыков опрятности) на фоне терапии церебролизином, массажа, коррекционных занятий (восстановительного обучения). В пользу терапевтической ремиссии данного состояния свидетельствует восстановление перечисленных функций, что не характерно для аутохтонного типа, при котором, как правило, не наблюдается восстановления ходьбы и столь значительной редукции ручных стереотипии, а также оживления в эмоциональной сфере, восстановления лепетной речи. Особенно обращает на себя внимание восстановление моторно-статических функций.
Читайте также: